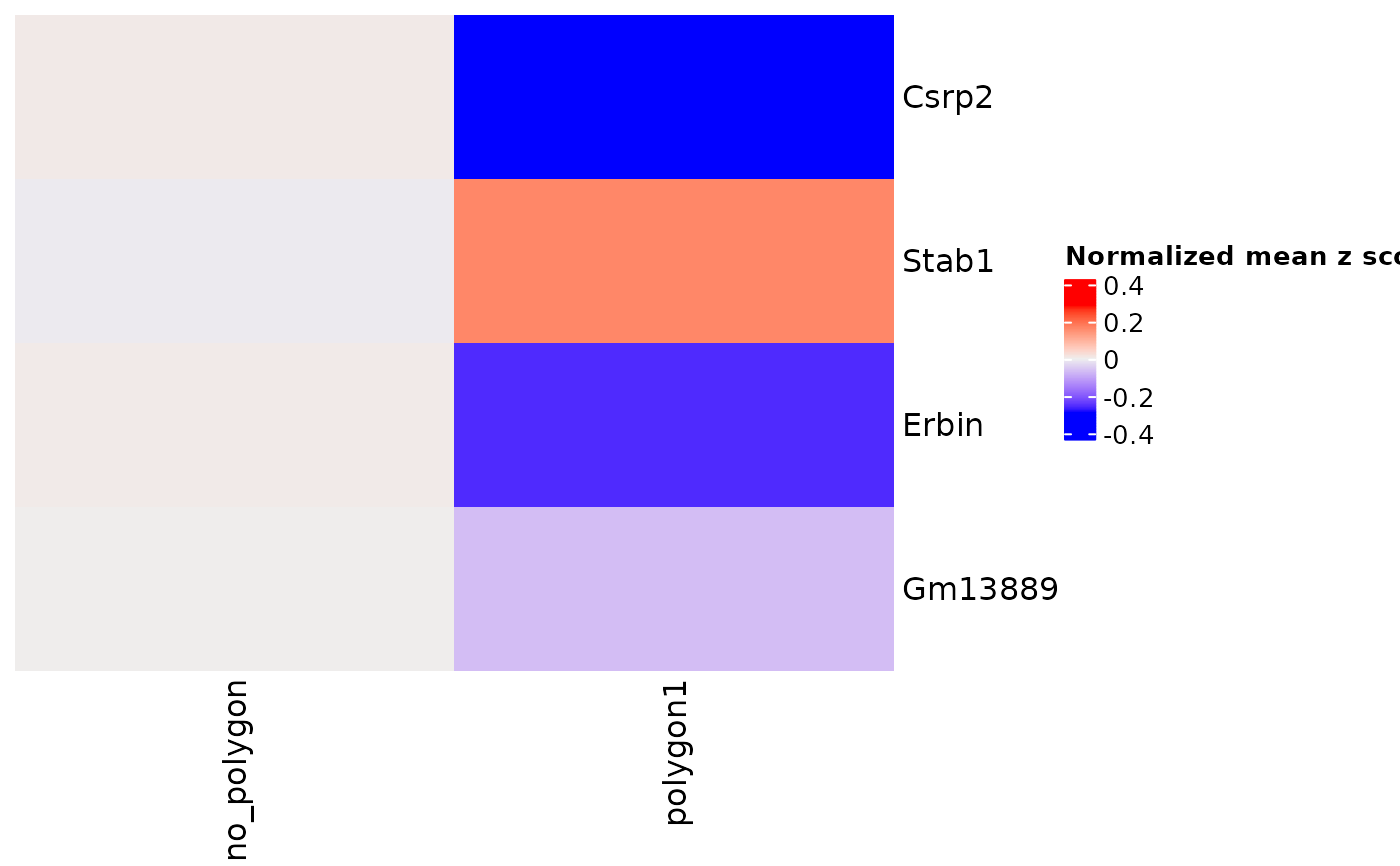
Compare gene expression between polygon areas
comparePolygonExpression(
gobject,
polygon_name = "selections",
spat_unit = "cell",
feat_type = "rna",
selected_feats = "top_genes",
expression_values = "normalized",
method = "scran",
...
)Arguments
- gobject
A Giotto object
- polygon_name
name of polygon selections
- spat_unit
spatial unit (e.g. "cell")
- feat_type
feature type (e.g. "rna", "dna", "protein")
- selected_feats
vector of selected features to plot
- expression_values
gene expression values to use ("normalized", "scaled", "custom")
- method
method to use to detect differentially expressed feats ("scran", "gini", "mast")
- ...
Arguments passed to Heatmap
Value
A ComplexHeatmap::Heatmap object
Examples
## Plot interactive polygons
g <- GiottoData::loadGiottoMini("visium")
#> 1. read Giotto object
#> 2. read Giotto feature information
#> 3. read Giotto spatial information
#> 3.1 read Giotto spatial shape information
#> 3.2 read Giotto spatial centroid information
#> 3.3 read Giotto spatial overlap information
#> 4. read Giotto image information
#> python already initialized in this session
#> active environment : '/usr/bin/python3'
#> python version : 3.12
my_polygon_coords <- data.frame(
poly_ID = rep("polygon1", 3),
sdimx = c(5477, 5959, 4720), sdimy = c(-4125, -2808, -5202)
)
## Add polygon coordinates to Giotto object
my_giotto_polygons <- createGiottoPolygonsFromDfr(my_polygon_coords,
name = "selections"
)
#> Selecting col "poly_ID" as poly_ID column
#> Selecting cols "sdimx" and "sdimy" as x and y respectively
g <- addGiottoPolygons(
gobject = g,
gpolygons = list(my_giotto_polygons)
)
## Add polygon cells
g <- addPolygonCells(g)
#>
#> These column names were already used: in_tissue nr_feats perc_feats total_expr
#> leiden_clus custom_leiden
#> and will be overwritten
comparePolygonExpression(g)
#> using 'Scran' to detect marker feats. If used in published
#> research, please cite: Lun ATL, McCarthy DJ, Marioni JC (2016).
#> 'A step-by-step workflow for low-level analysis of single-cell RNA-seq
#> data with Bioconductor.'
#> F1000Res., 5, 2122. doi: 10.12688/f1000research.9501.2.
#> start with cluster no_polygonstart with cluster polygon1