Open-ST Mouse Hippocampus
Joselyn C. Chávez Fuentes, Caleb Hahn, Brandon Tung
Source:vignettes/openst_mouse_hippocampus.Rmd
openst_mouse_hippocampus.Rmd1 Dataset explanation
Open-ST is an open-source spatial transcriptomics method with efficient whole-transcriptome capture at sub-cellular resolution (0.6 μm) at low cost (<150 Euro library preparation per 12 mm²). You can find more information in the original article.
The Adult mouse hippocampus data to run this tutorial can be found here. Download the file adult_mouse_hippocampus_by_cell.h5ad.tar.gz and untar it to run this tutorial.
2 Set up Giotto Environment
# Ensure Giotto Suite is installed.
if(!"Giotto" %in% installed.packages()) {
pak::pkg_install("drieslab/Giotto")
}
# Ensure the Python environment for Giotto has been installed.
genv_exists <- Giotto::checkGiottoEnvironment()
if(!genv_exists){
# The following command need only be run once to install the Giotto environment.
Giotto::installGiottoEnvironment()
}3 Create Giotto object and visualize
library(Giotto)
# 1. set working directory
results_folder <- "path/to/results"
# Optional: Specify a path to a Python executable within a conda or miniconda
# environment. If set to NULL (default), the Python executable within the previously
# installed Giotto environment will be used.
python_path <- NULL # alternatively, "/local/python/path/python" if desired.
## provide path to the data folder
data_path <- "/path/to/data/"
## create the object directly from the h5ad file
giotto_object <- anndataToGiotto(file.path(data_path, "openst_demo_adult_mouse_by_cell.h5ad"),
python_path = python_path)
## update instructions
instructions(giotto_object, "save_plot") <- TRUE
instructions(giotto_object, "save_dir") <- results_folder
instructions(giotto_object, "show_plot") <- FALSE
instructions(giotto_object, "return_plot") <- FALSE
## The cell with the identifier 0 will have all the transcripts belonging to the background. Therefore, make sure to omit it from the dataset.
cell_metadata <- pDataDT(giotto_object)
cell_metadata <- cell_metadata[cell_metadata$cell_ID != "0",]
giotto_object <- subsetGiotto(giotto_object,
cell_ids = cell_metadata$cell_ID)
## show plot
spatPlot2D(gobject = giotto_object,
point_size = 1)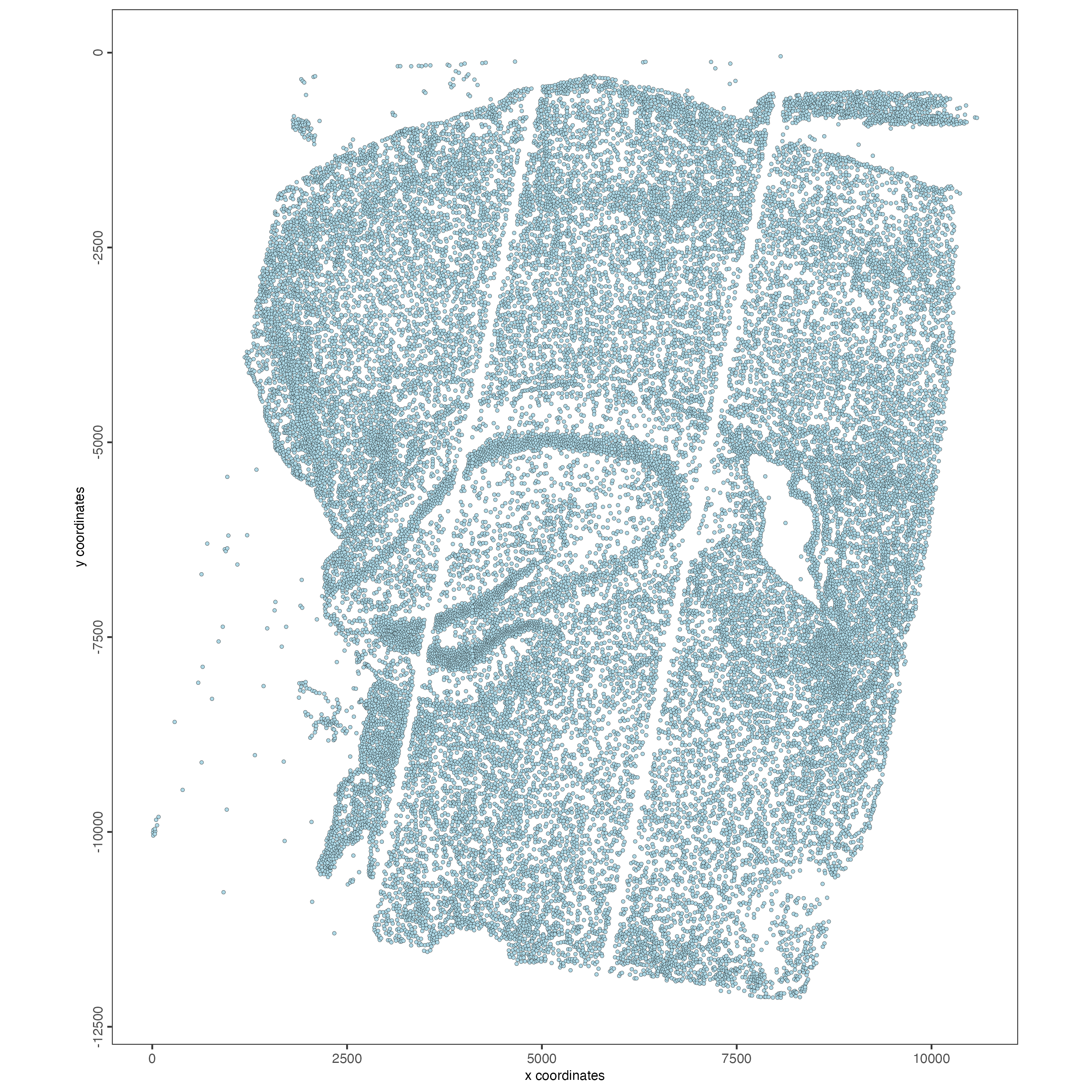
4 QC
giotto_object_statistics <- addStatistics(giotto_object,
expression_values = "raw")
filterDistributions(gobject = giotto_object_statistics,
detection = "cells",
nr_bins = 100)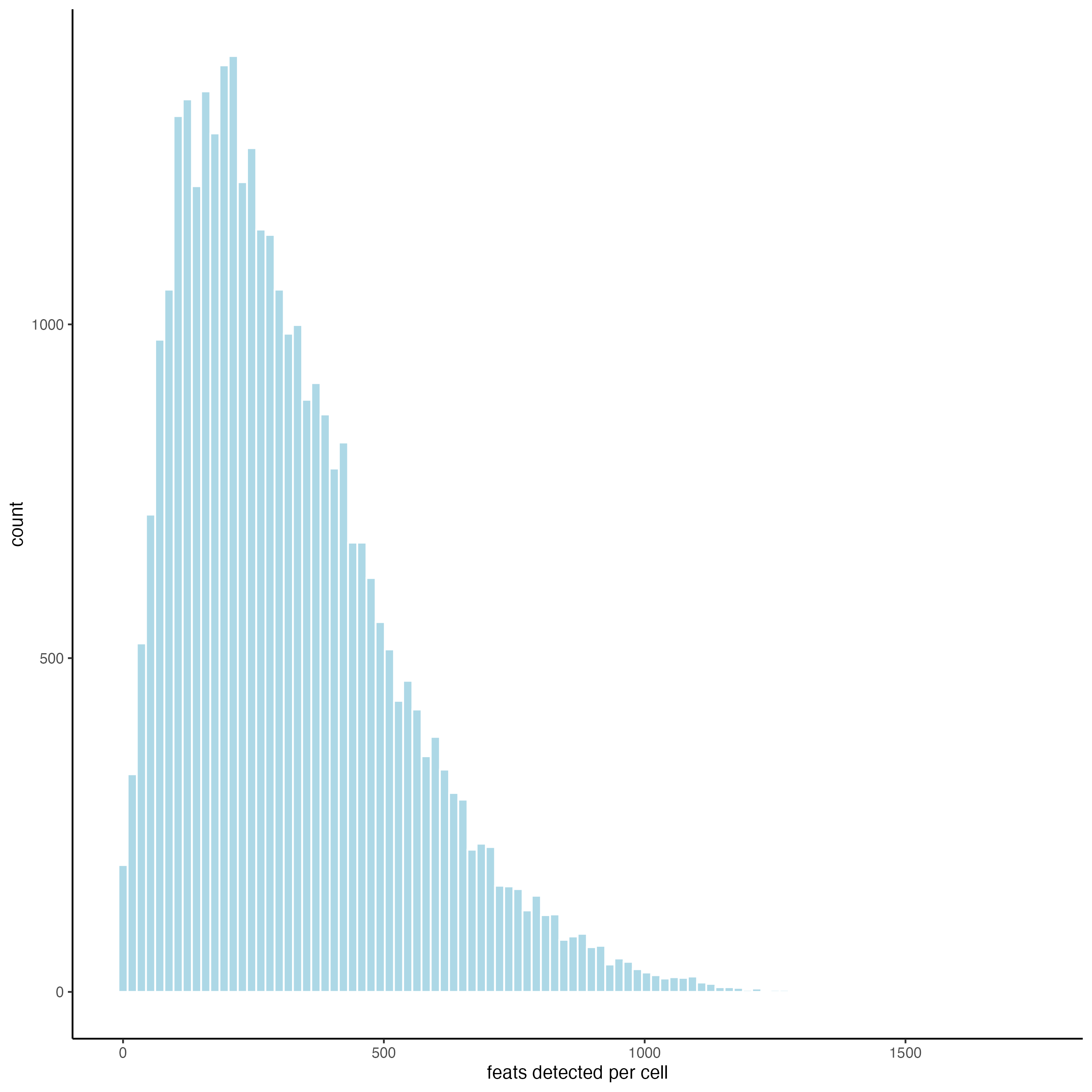
filterDistributions(gobject = giotto_object_statistics,
detection = "feats",
nr_bins = 100)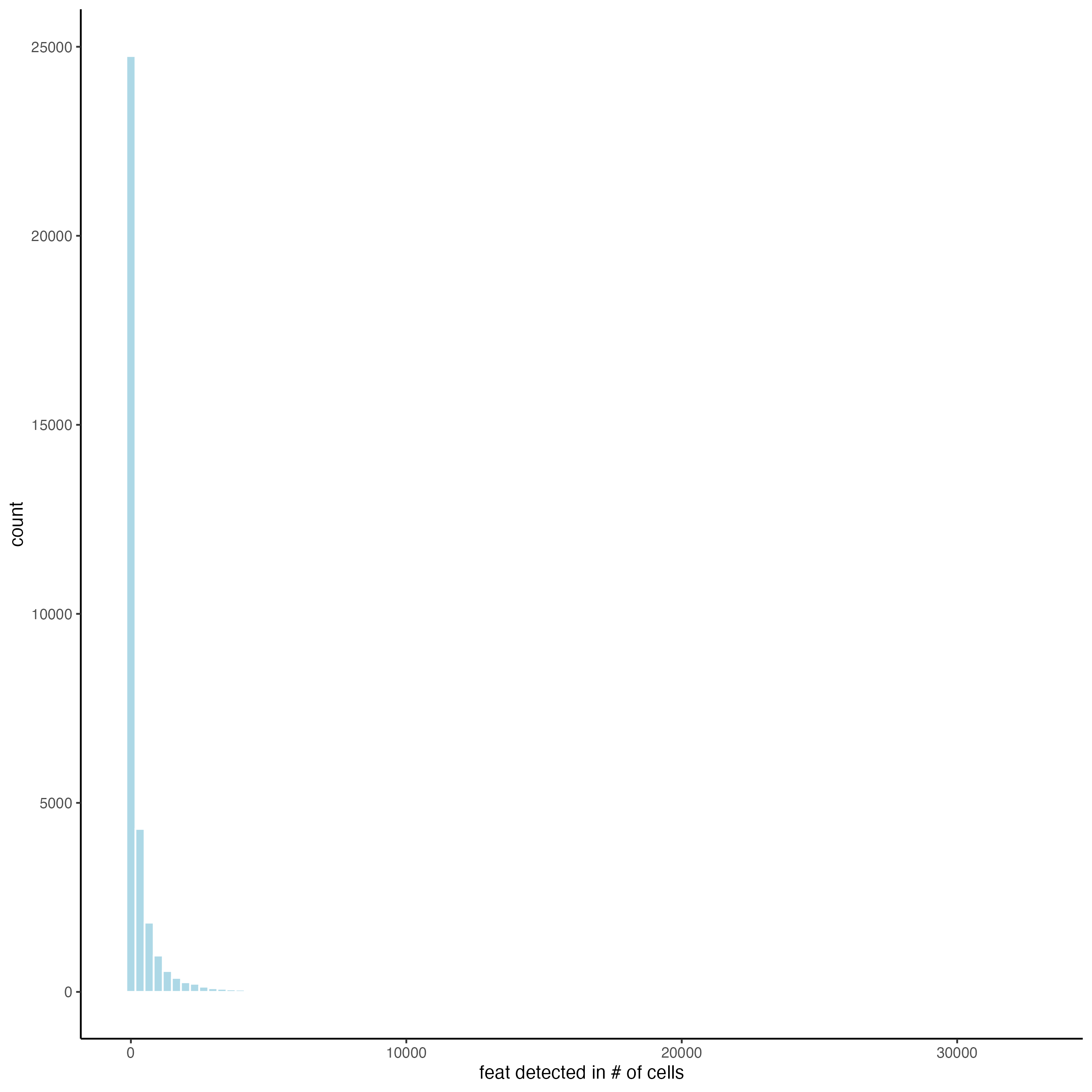
5 Process Giotto Object
## filter
giotto_object <- filterGiotto(gobject = giotto_object,
expression_threshold = 1,
feat_det_in_min_cells = 25,
min_det_feats_per_cell = 100,
verbose = TRUE)
## normalize
giotto_object <- normalizeGiotto(gobject = giotto_object,
scalefactor = 6000,
verbose = TRUE)
## add gene & cell statistics
giotto_object <- addStatistics(gobject = giotto_object)
## visualize
spatPlot2D(gobject = giotto_object,
cell_color = "nr_feats",
color_as_factor = FALSE,
point_size = 1,
gradient_limits = c(0, 1000))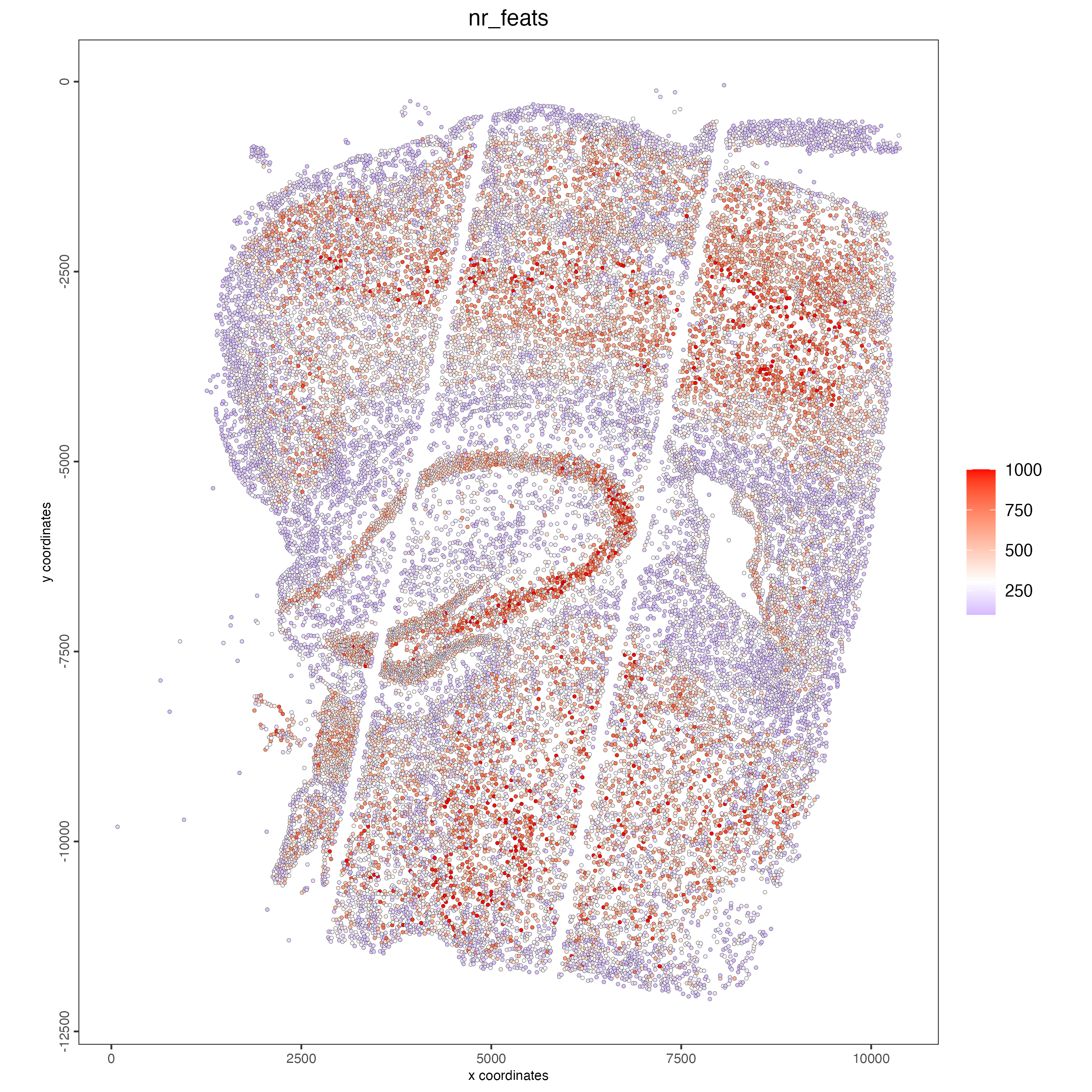
6 Dimension Reduction
## run PCA on expression values
giotto_object <- runPCA(gobject = giotto_object)
screePlot(giotto_object,
ncp = 30)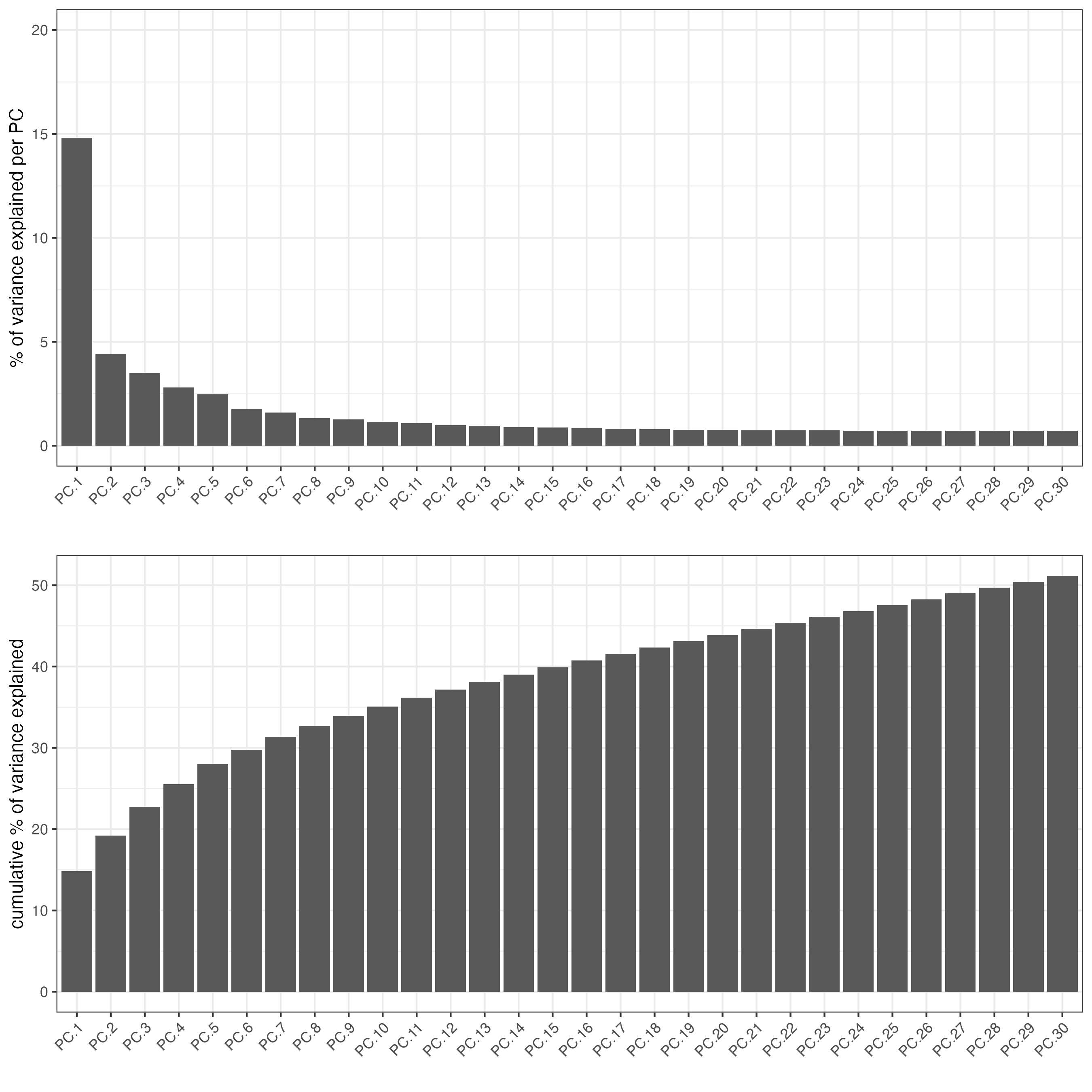
plotPCA(gobject = giotto_object,
point_size = 1)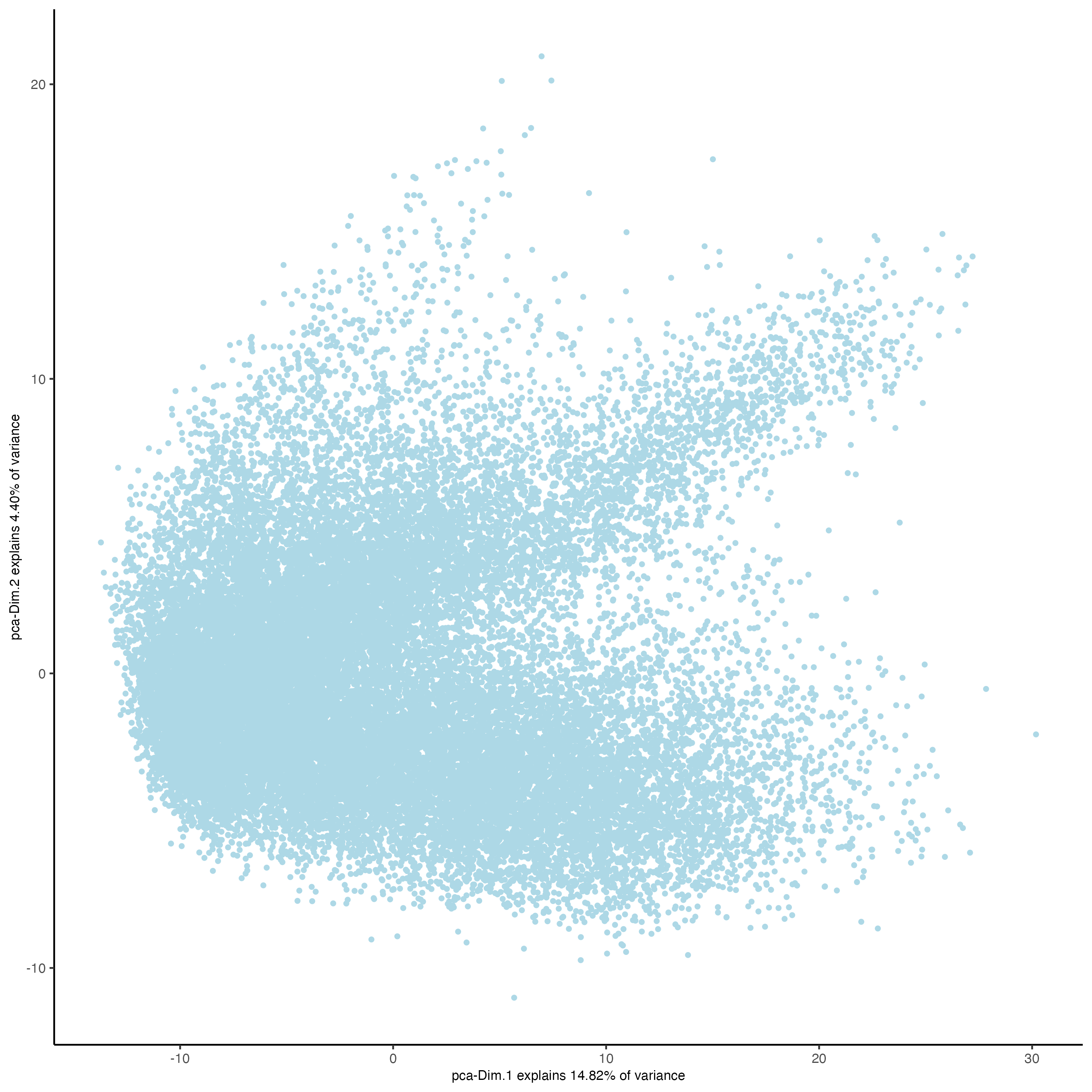
## run UMAP and tSNE on PCA space (default)
giotto_object <- runUMAP(giotto_object,
dimensions_to_use = 1:10)
giotto_object <- runtSNE(giotto_object,
dimensions_to_use = 1:10)7 Clustering
## sNN network (default)
giotto_object <- createNearestNetwork(gobject = giotto_object,
dimensions_to_use = 1:10,
k = 15)
## Leiden clustering
giotto_object <- doLeidenCluster(gobject = giotto_object,
resolution = 0.5,
n_iterations = 1000)
plotUMAP(gobject = giotto_object,
cell_color = "leiden_clus",
point_size = 2)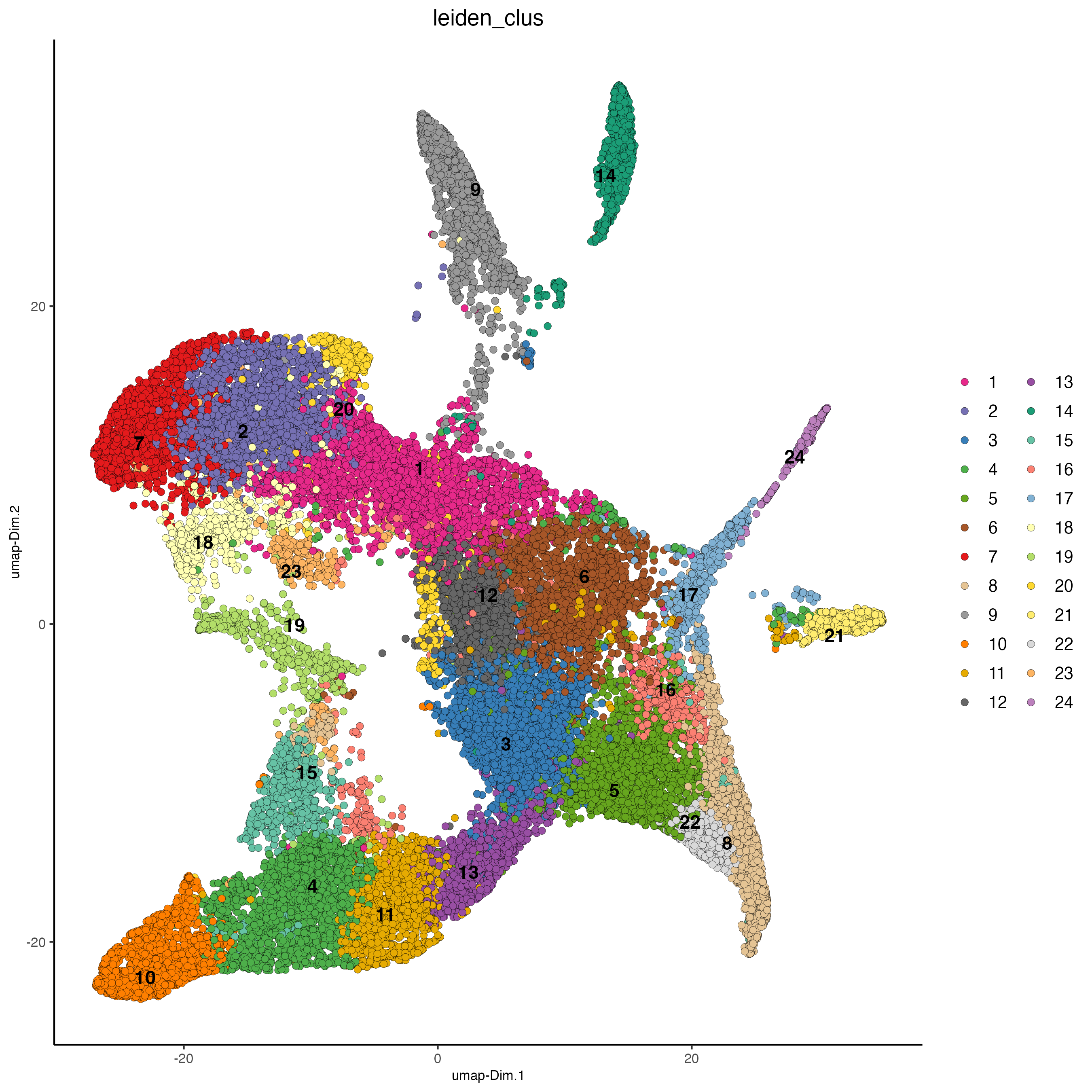
plotTSNE(gobject = giotto_object,
cell_color = "leiden_clus",
point_size = 2)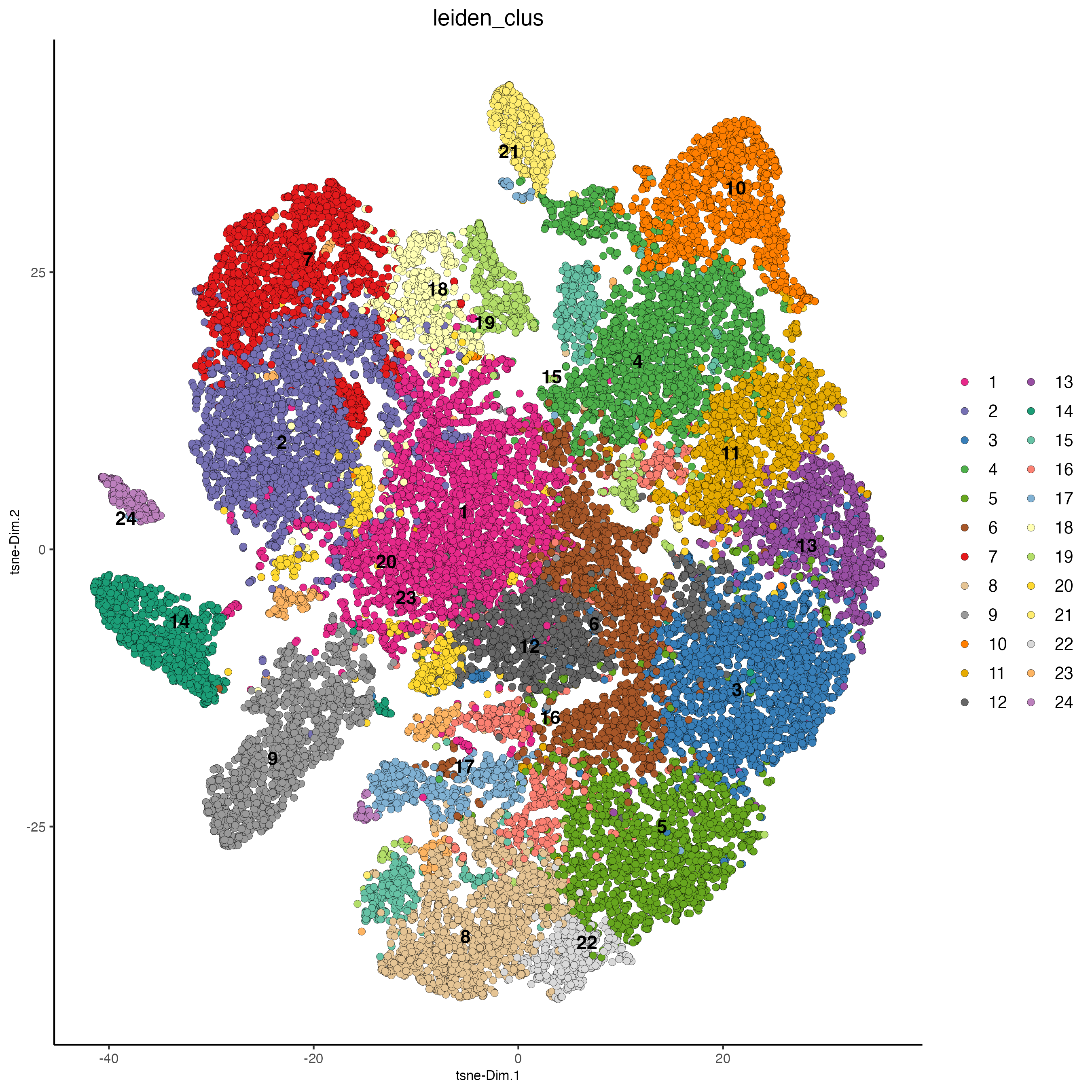
# spatial plot
spatPlot2D(gobject = giotto_object,
cell_color = "leiden_clus",
point_size = 1)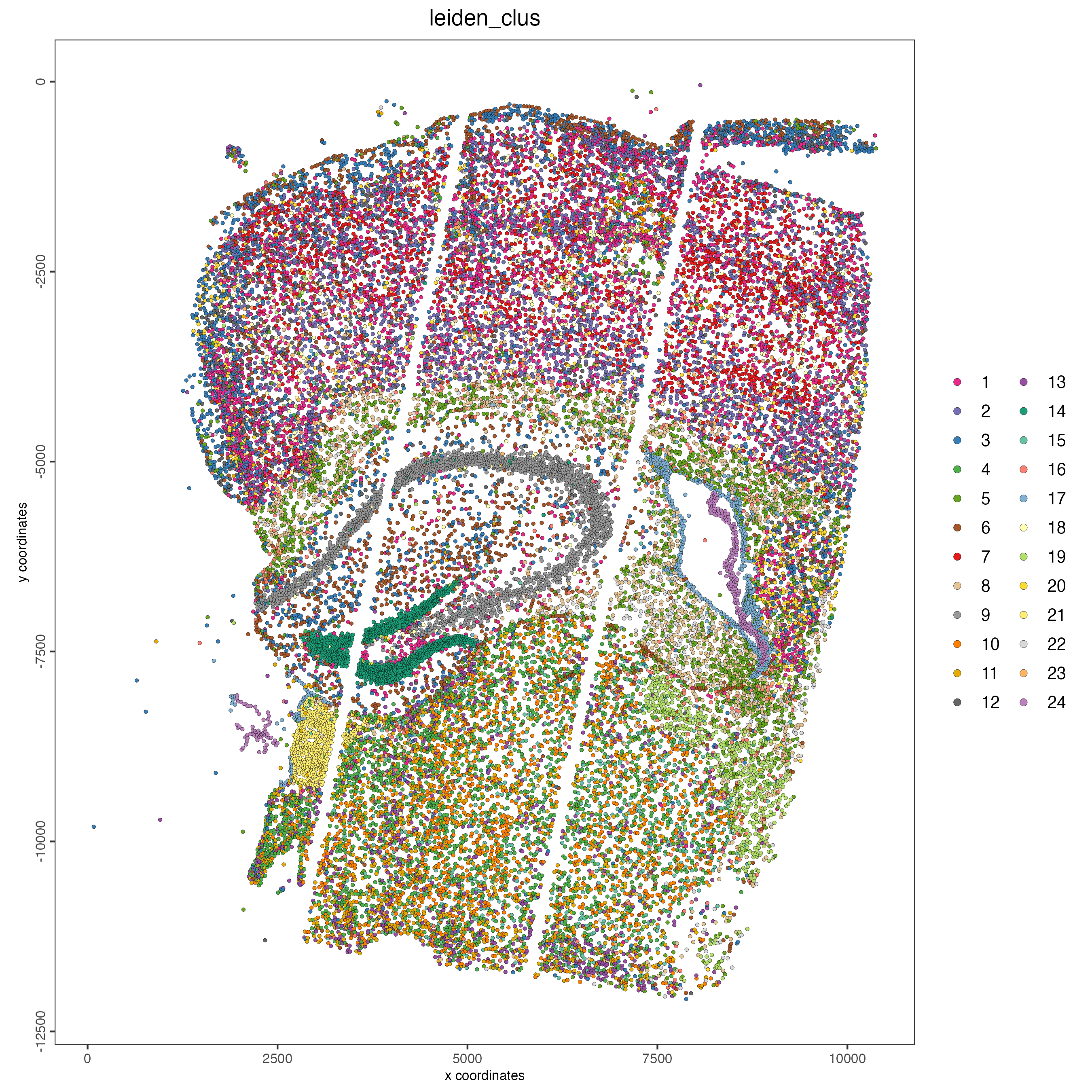
8 Spatial network
giotto_object <- createSpatialNetwork(gobject = giotto_object,
method = "kNN",
k = 5,
maximum_distance_knn = 400,
name = "spatial_network")
spatPlot2D(gobject = giotto_object,
show_network = TRUE,
network_color = "blue",
spatial_network_name = "spatial_network",
point_size = 1)
9 Spatial Genes
## rank binarization
ranktest <- binSpect(giotto_object,
bin_method = "rank",
calc_hub = TRUE,
hub_min_int = 5,
spatial_network_name = "spatial_network")
spatFeatPlot2D(giotto_object,
expression_values = "scaled",
feats = ranktest$feats[1:6],
cow_n_col = 2,
point_size = 1)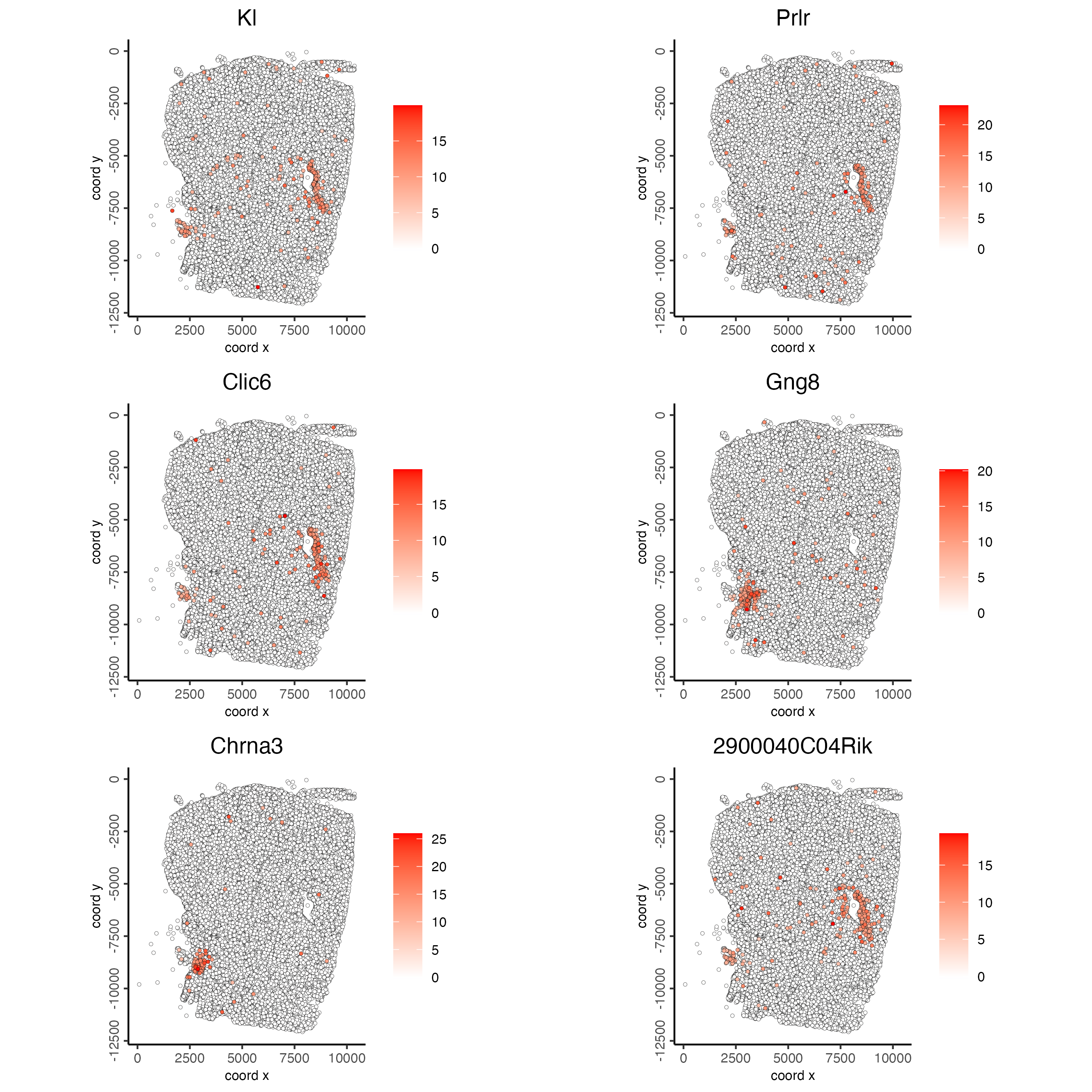
10 Spatial Co-Expression modules
# cluster the top 500 spatial genes into 10 clusters
my_spatial_genes <- ranktest[1:500,]$feats
# here we use existing detectSpatialCorGenes function to calculate pairwise distances between genes (but set network_smoothing=0 to use default clustering)
spat_cor_netw_DT <- detectSpatialCorFeats(giotto_object,
method = "network",
spatial_network_name = "spatial_network",
subset_feats = my_spatial_genes)
# 2. identify most similar spatially correlated genes for one gene
top10_genes <- showSpatialCorFeats(spat_cor_netw_DT,
feats = "Kl",
show_top_feats = 10)
spatFeatPlot2D(giotto_object,
expression_values = "scaled",
feats = top10_genes$variable[1:4],
point_size = 1)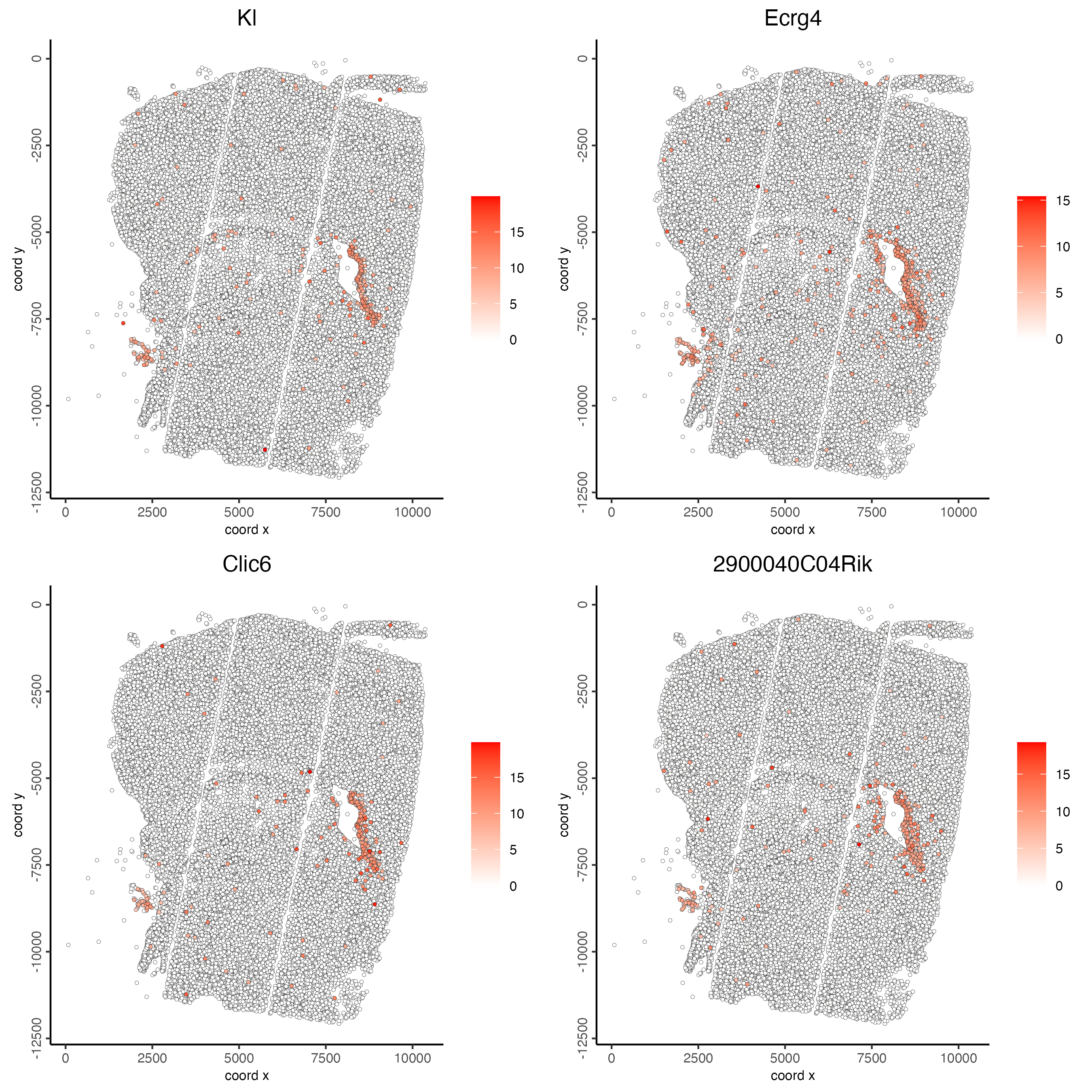
# cluster spatial genes
spat_cor_netw_DT <- clusterSpatialCorFeats(spat_cor_netw_DT,
name = "spat_netw_clus",
k = 6)
# visualize clusters
heatmSpatialCorFeats(giotto_object,
spatCorObject = spat_cor_netw_DT,
use_clus_name = "spat_netw_clus",
heatmap_legend_param = list(title = NULL))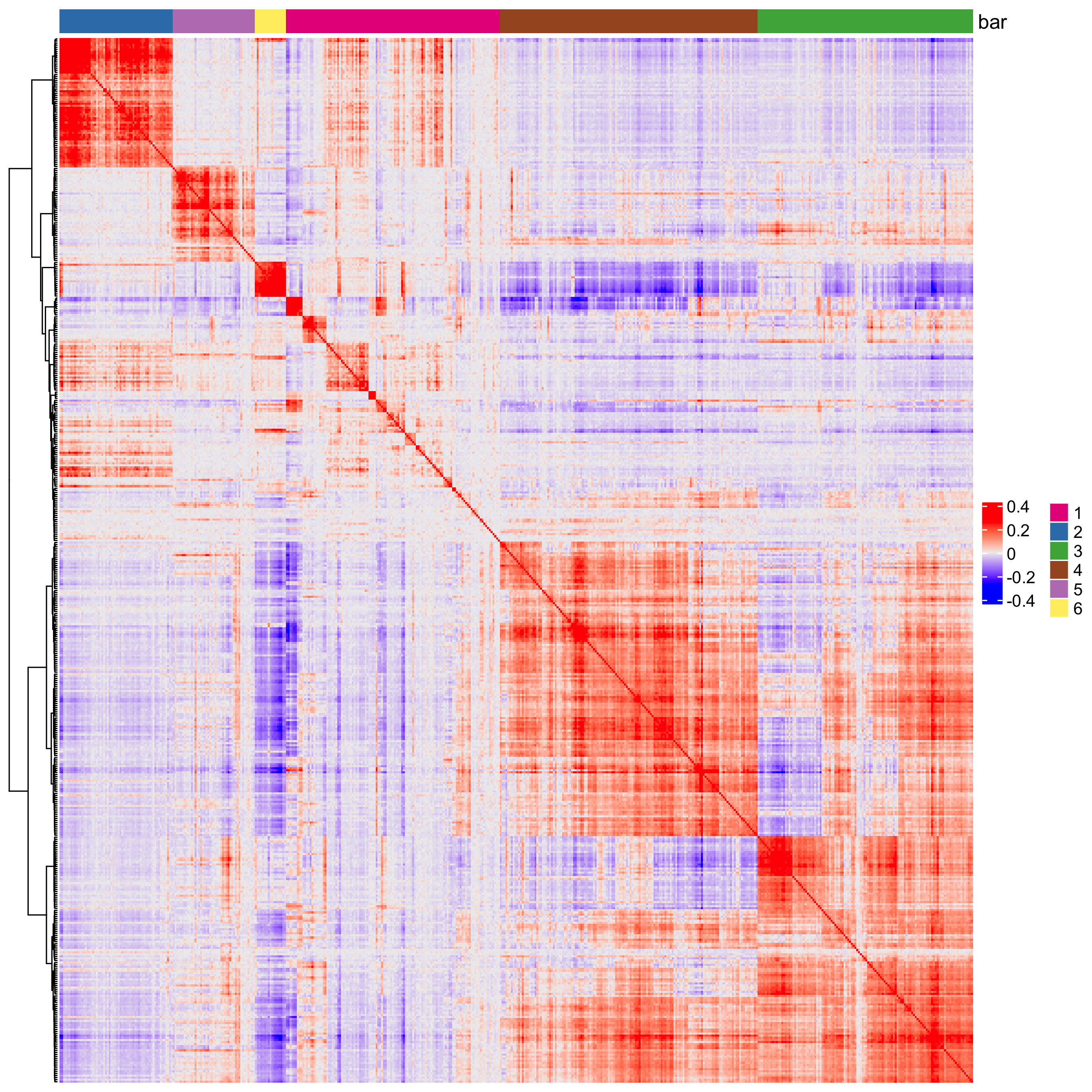
# 4. rank spatial correlated clusters and show genes for selected clusters
netw_ranks <- rankSpatialCorGroups(giotto_object,
spatCorObject = spat_cor_netw_DT,
use_clus_name = "spat_netw_clus")
top_netw_spat_cluster <- showSpatialCorFeats(spat_cor_netw_DT,
use_clus_name = "spat_netw_clus",
selected_clusters = 6,
show_top_feats = 1)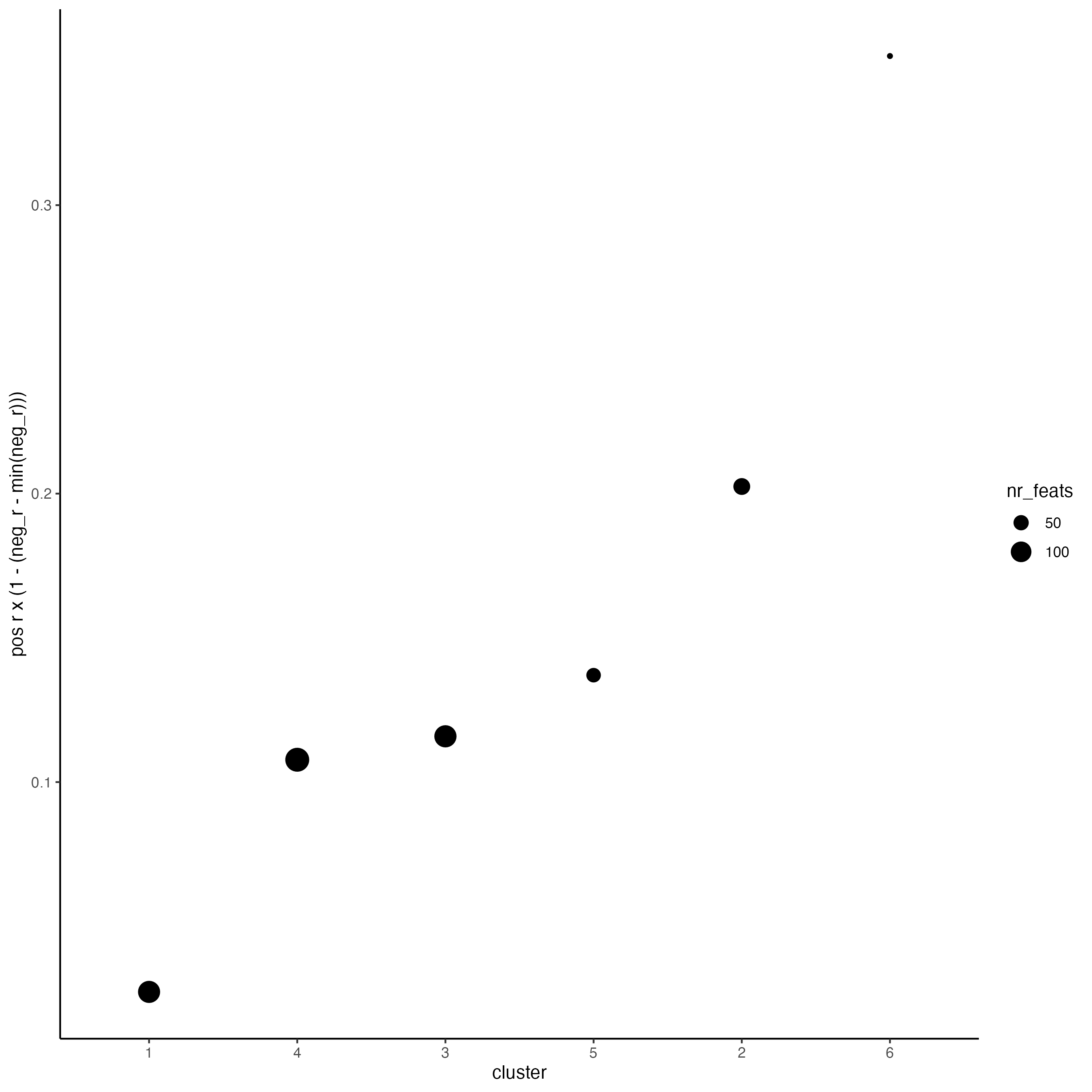
# 5. create metagene enrichment score for clusters
cluster_genes_DT <- showSpatialCorFeats(spat_cor_netw_DT,
use_clus_name = "spat_netw_clus",
show_top_feats = 1)
cluster_genes <- cluster_genes_DT$clus
names(cluster_genes) <- cluster_genes_DT$feat_ID
giotto_object <- createMetafeats(giotto_object,
feat_clusters = cluster_genes,
name = "cluster_metagene")
spatCellPlot(giotto_object,
spat_enr_names = "cluster_metagene",
cell_annotation_values = netw_ranks$clusters,
point_size = 0.3,
cow_n_col = 2,
gradient_limits = c(-2,2))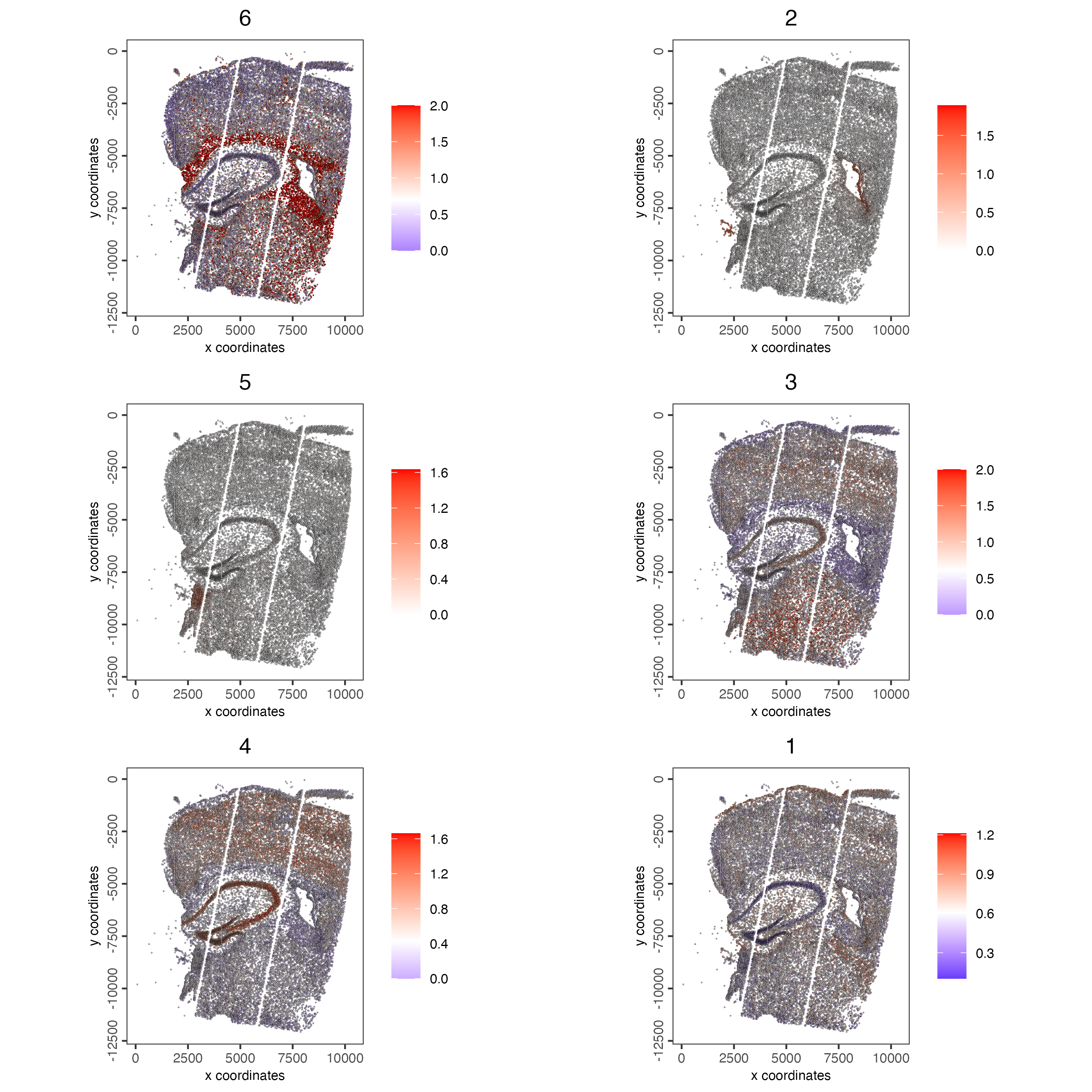
11 Spatially informed clusters
# top 30 genes per spatial co-expression cluster
table(spat_cor_netw_DT$cor_clusters$spat_netw_clus)
coexpr_dt <- data.table::data.table(
genes = names(spat_cor_netw_DT$cor_clusters$spat_netw_clus),
cluster = spat_cor_netw_DT$cor_clusters$spat_netw_clus)
data.table::setorder(coexpr_dt, cluster)
top30_coexpr_dt <- coexpr_dt[, head(.SD, 30), by = cluster]
my_spatial_genes <- top30_coexpr_dt$genes
giotto_object <- runPCA(gobject = giotto_object,
feats_to_use = my_spatial_genes,
name = "custom_pca")
giotto_object <- runUMAP(giotto_object,
dim_reduction_name = "custom_pca",
dimensions_to_use = 1:20,
name = "custom_umap")
giotto_object <- createNearestNetwork(gobject = giotto_object,
dim_reduction_name = "custom_pca",
dimensions_to_use = 1:20,
k = 5,
name = "custom_NN")
giotto_object <- doLeidenCluster(gobject = giotto_object,
network_name = "custom_NN",
resolution = 0.1,
n_iterations = 1000,
name = "custom_leiden")
cell_metadata <- pDataDT(giotto_object)
cell_clusters <- unique(cell_metadata$custom_leiden)
giotto_colors <- getDistinctColors(length(cell_clusters))
names(giotto_colors) <- cell_clusters
spatPlot2D(giotto_object,
cell_color = "custom_leiden",
cell_color_code = giotto_colors,
point_size = 1)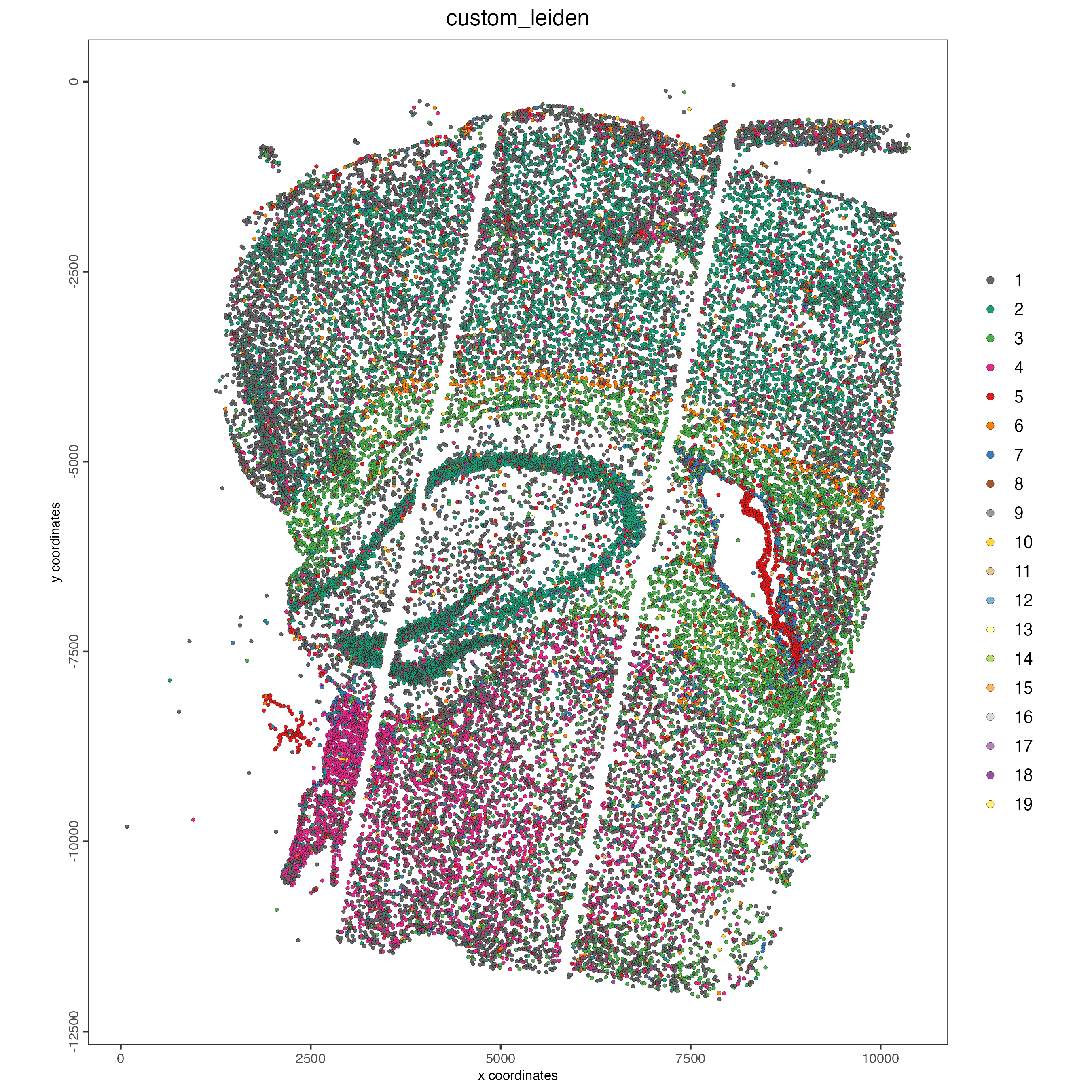
12 Session info
R version 4.4.0 (2024-04-24)
Platform: x86_64-apple-darwin20
Running under: macOS Sonoma 14.6.1
Matrix products: default
BLAS: /System/Library/Frameworks/Accelerate.framework/Versions/A/Frameworks/vecLib.framework/Versions/A/libBLAS.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.4-x86_64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: America/New_York
tzcode source: internal
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] Giotto_4.1.1 GiottoClass_0.3.5
loaded via a namespace (and not attached):
[1] colorRamp2_0.1.0 deldir_2.0-4 rlang_1.1.4
[4] magrittr_2.0.3 clue_0.3-65 GetoptLong_1.0.5
[7] RcppAnnoy_0.0.22 GiottoUtils_0.1.11 matrixStats_1.3.0
[10] compiler_4.4.0 png_0.1-8 systemfonts_1.1.0
[13] vctrs_0.6.5 reshape2_1.4.4 stringr_1.5.1
[16] shape_1.4.6.1 pkgconfig_2.0.3 SpatialExperiment_1.14.0
[19] crayon_1.5.3 fastmap_1.2.0 backports_1.5.0
[22] magick_2.8.4 XVector_0.44.0 labeling_0.4.3
[25] utf8_1.2.4 rmarkdown_2.28 UCSC.utils_1.0.0
[28] ragg_1.3.2 purrr_1.0.2 xfun_0.47
[31] beachmat_2.20.0 zlibbioc_1.50.0 GenomeInfoDb_1.40.1
[34] jsonlite_1.8.8 DelayedArray_0.30.1 BiocParallel_1.38.0
[37] terra_1.7-78 cluster_2.1.6 irlba_2.3.5.1
[40] parallel_4.4.0 R6_2.5.1 stringi_1.8.4
[43] RColorBrewer_1.1-3 reticulate_1.38.0 GenomicRanges_1.56.1
[46] scattermore_1.2 iterators_1.0.14 Rcpp_1.0.13
[49] SummarizedExperiment_1.34.0 knitr_1.48 R.utils_2.12.3
[52] IRanges_2.38.1 Matrix_1.7-0 igraph_2.0.3
[55] tidyselect_1.2.1 rstudioapi_0.16.0 abind_1.4-5
[58] yaml_2.3.10 doParallel_1.0.17 codetools_0.2-20
[61] lattice_0.22-6 tibble_3.2.1 plyr_1.8.9
[64] Biobase_2.64.0 withr_3.0.1 Rtsne_0.17
[67] evaluate_0.24.0 circlize_0.4.16 pillar_1.9.0
[70] MatrixGenerics_1.16.0 foreach_1.5.2 checkmate_2.3.2
[73] stats4_4.4.0 plotly_4.10.4 generics_0.1.3
[76] dbscan_1.2-0 sp_2.1-4 S4Vectors_0.42.1
[79] ggplot2_3.5.1 munsell_0.5.1 scales_1.3.0
[82] gtools_3.9.5 glue_1.7.0 lazyeval_0.2.2
[85] tools_4.4.0 GiottoVisuals_0.2.5 data.table_1.15.4
[88] ScaledMatrix_1.12.0 Cairo_1.6-2 cowplot_1.1.3
[91] grid_4.4.0 tidyr_1.3.1 colorspace_2.1-1
[94] SingleCellExperiment_1.26.0 GenomeInfoDbData_1.2.12 BiocSingular_1.20.0
[97] rsvd_1.0.5 cli_3.6.3 textshaping_0.4.0
[100] fansi_1.0.6 S4Arrays_1.4.1 viridisLite_0.4.2
[103] ComplexHeatmap_2.20.0 dplyr_1.1.4 uwot_0.2.2
[106] gtable_0.3.5 R.methodsS3_1.8.2 digest_0.6.37
[109] BiocGenerics_0.50.0 SparseArray_1.4.8 ggrepel_0.9.5
[112] rjson_0.2.22 htmlwidgets_1.6.4 farver_2.1.2
[115] htmltools_0.5.8.1 R.oo_1.26.0 lifecycle_1.0.4
[118] httr_1.4.7 GlobalOptions_0.1.2