1 Dataset explanation
The CODEX data to run this tutorial can be found here. Alternatively you can use GiottoData::getSpatialDataset to automatically download this dataset like we do in this example.
Goltsev et al. created a multiplexed datasets of normal and lupus (MRL/lpr) murine spleens using CODEX technique. The dataset consists of 30 protein markers from 734,101 single cells. In this tutorial, 83,787 cells from sample “BALBc-3” were selected for the analysis.
2 Set up Giotto environment
# Ensure Giotto Suite is installed.
if(!"Giotto" %in% installed.packages()) {
pak::pkg_install("drieslab/Giotto")
}
# Ensure GiottoData, a small, helper module for tutorials, is installed.
if(!"GiottoData" %in% installed.packages()) {
pak::pkg_install("drieslab/GiottoData")
}
# Ensure the Python environment for Giotto has been installed.
genv_exists <- Giotto::checkGiottoEnvironment()
if(!genv_exists){
# The following command need only be run once to install the Giotto environment.
Giotto::installGiottoEnvironment()
}3 Giotto global instructions and preparations
library(Giotto)
library(GiottoData)
# 1. set working directory
results_folder <- "/path/to/results/"
# Optional: Specify a path to a Python executable within a conda or miniconda
# environment. If set to NULL (default), the Python executable within the previously
# installed Giotto environment will be used.
python_path <- NULL # alternatively, "/local/python/path/python" if desired.
# download data to working directory
# use method = "wget" if wget is available. This should be much faster.
# if you run into authentication issues with wgeTRUE, then add " extra = "--no-check-certificate" "
getSpatialDataset(dataset = "codex_spleen",
directory = results_folder,
method = "wget")# 1. (optional) set Giotto instructions
instructions <- createGiottoInstructions(save_plot = TRUE,
show_plot = FALSE,
return_plot = FALSE
save_dir = results_folder,
python_path = python_path)
# 2. create giotto object from provided paths ####
expr_path <- paste0(results_folder, "codex_BALBc_3_expression.txt.gz")
loc_path <- paste0(results_folder, "codex_BALBc_3_coord.txt")
meta_path <- paste0(results_folder, "codex_BALBc_3_annotation.txt")4 Create Giotto object & process data
# read in data information
# expression info
codex_expression <- readExprMatrix(expr_path, transpose = FALSE)
# cell coordinate info
codex_locations <- data.table::fread(loc_path)
# metadata
codex_metadata <- data.table::fread(meta_path)
## stitch x.y tile coordinates to global coordinates
xtilespan <- 1344
ytilespan <- 1008
# TODO: expand the documentation and input format of stitchTileCoordinates. Probably not enough information for new users.
stitch_file <- stitchTileCoordinates(location_file = codex_metadata,
Xtilespan = xtilespan,
Ytilespan = ytilespan)
codex_locations <- stitch_file[,.(Xcoord, Ycoord)]
# create Giotto object
codex_test <- createGiottoObject(expression = codex_expression,
spatial_locs = codex_locations,
instructions = instructions)
codex_metadata$cell_ID <- as.character(codex_metadata$cellID)
codex_test <- addCellMetadata(codex_tesTRUE, new_metadata = codex_metadata,
by_column = TRUE,
column_cell_ID = "cell_ID")
# subset Giotto object
cell_metadata <- pDataDT(codex_test)
cell_IDs_to_keep <- cell_metadata[Imaging_phenotype_cell_type != "dirt" & Imaging_phenotype_cell_type != "noid" & Imaging_phenotype_cell_type != "capsule",]$cell_ID
codex_test <- subsetGiotto(codex_tesTRUE,
cell_ids = cell_IDs_to_keep)
## filter
codex_test <- filterGiotto(gobject = codex_tesTRUE,
expression_threshold = 1,
feat_det_in_min_cells = 10,
min_det_feats_per_cell = 2,
expression_values = "raw",
verbose = TRUE)
codex_test <- normalizeGiotto(gobject = codex_tesTRUE,
scalefactor = 6000,
verbose = TRUE,
log_norm = FALSE,
library_size_norm = FALSE,
scale_feats = FALSE,
scale_cells = TRUE)
## add gene & cell statistics
codex_test <- addStatistics(gobject = codex_tesTRUE,
expression_values = "normalized")
## adjust expression matrix for technical or known variables
codex_test <- adjustGiottoMatrix(gobject = codex_tesTRUE,
expression_values = "normalized",
batch_columns = "sample_Xtile_Ytile",
covariate_columns = NULL,
return_gobject = TRUE,
update_slot = "custom")
## visualize
spatPlot(gobject = codex_tesTRUE,
point_size = 0.1,
coord_fix_ratio = NULL,
point_shape = "no_border",
save_param = list(save_name = "2_a_spatPlot"))
Show different regions of the dataset
spatPlot(gobject = codex_tesTRUE,
point_size = 0.2,
coord_fix_ratio = 1,
cell_color = "sample_Xtile_Ytile",
legend_symbol_size = 3,
legend_text = 5,
save_param = list(save_name = "2_b_spatPlot"))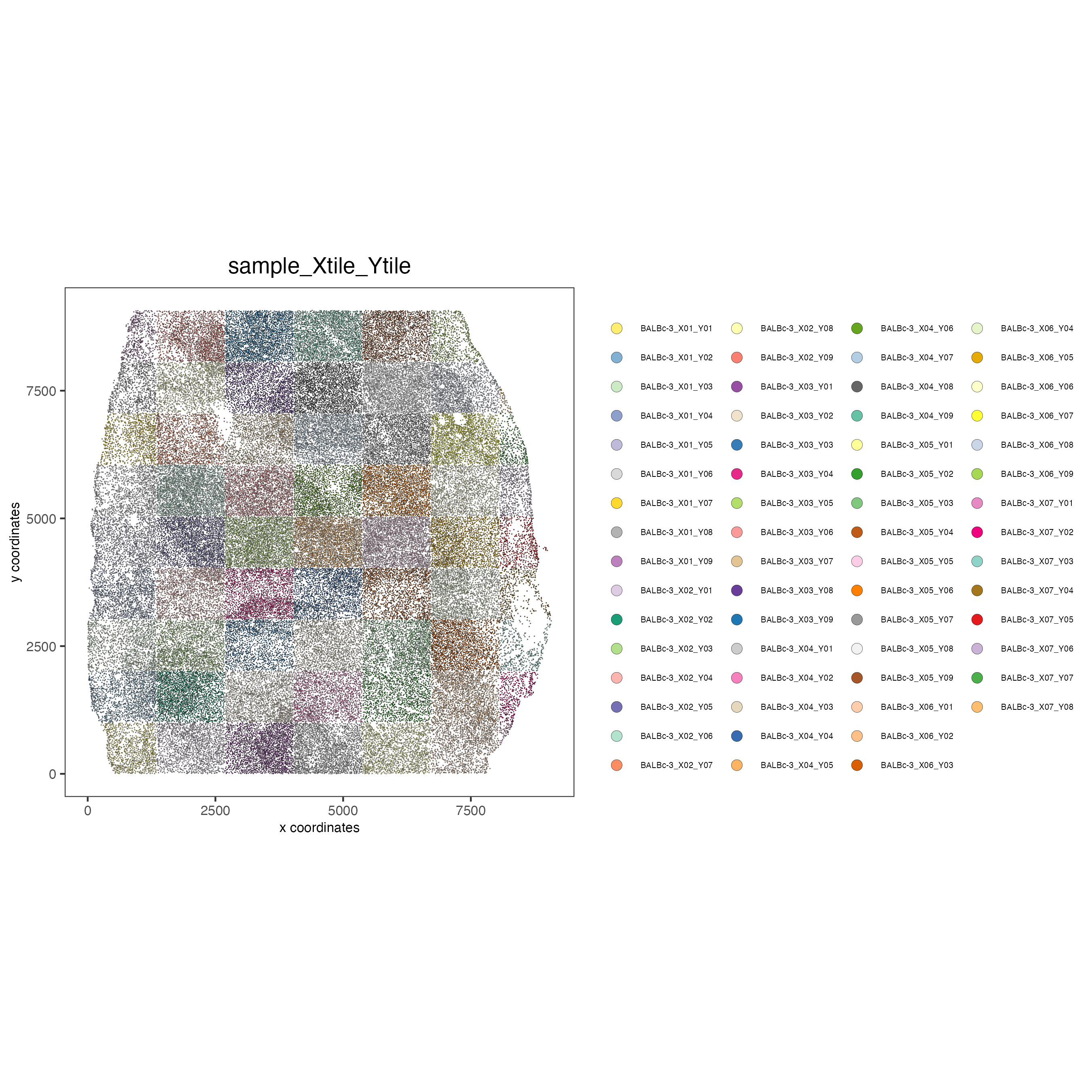
5 Dimension reduction
# use all Abs
# PCA
codex_test <- runPCA(gobject = codex_tesTRUE,
expression_values = "normalized",
scale_unit = TRUE,
method = "factominer")
signPCA(codex_tesTRUE,
scale_unit = TRUE,
scree_ylim = c(0, 3),
save_param = list(save_name = "3_a_spatPlot"))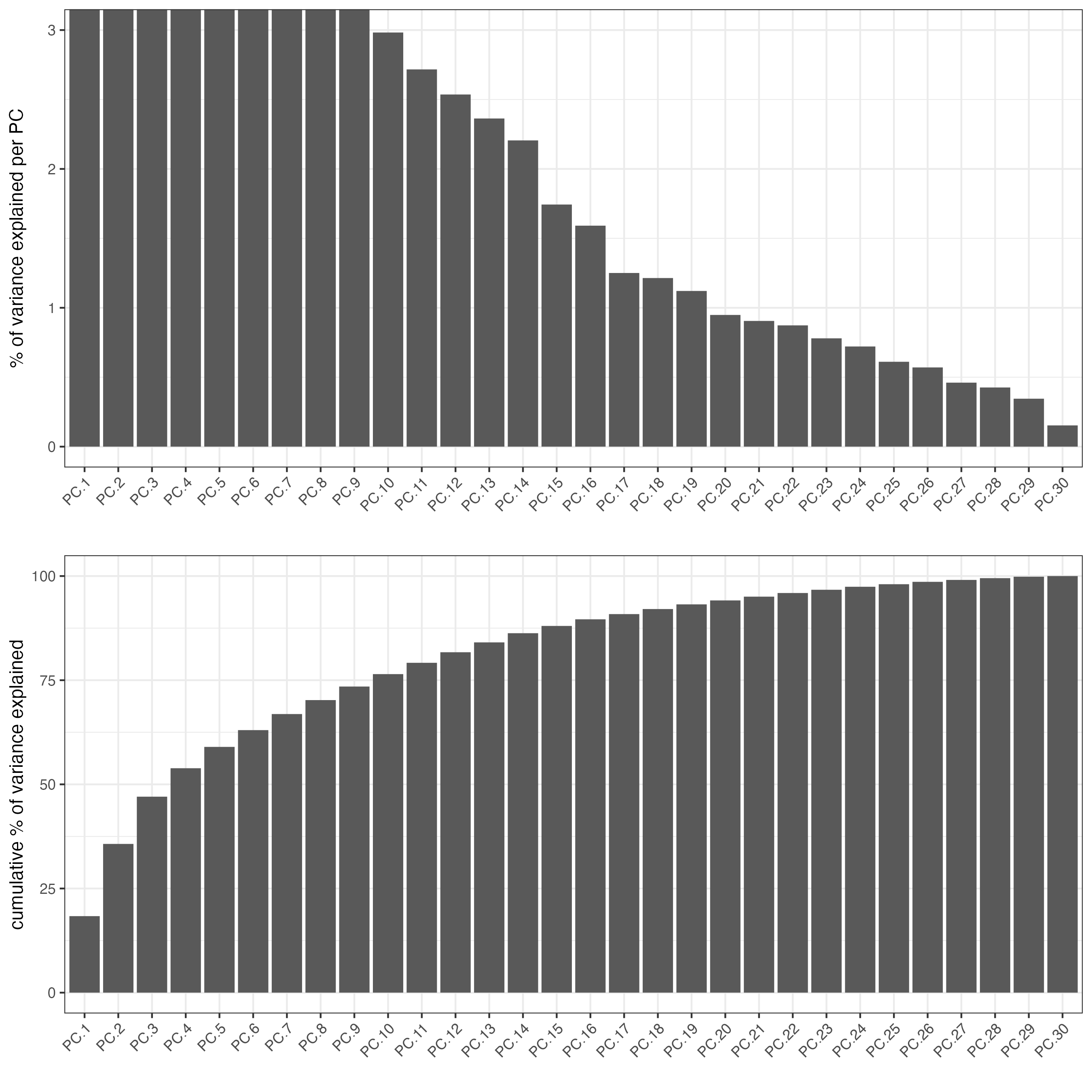
plotPCA(gobject = codex_tesTRUE,
point_shape = "no_border",
point_size = 0.2,
save_param = list(save_name = "3_b_PCA"))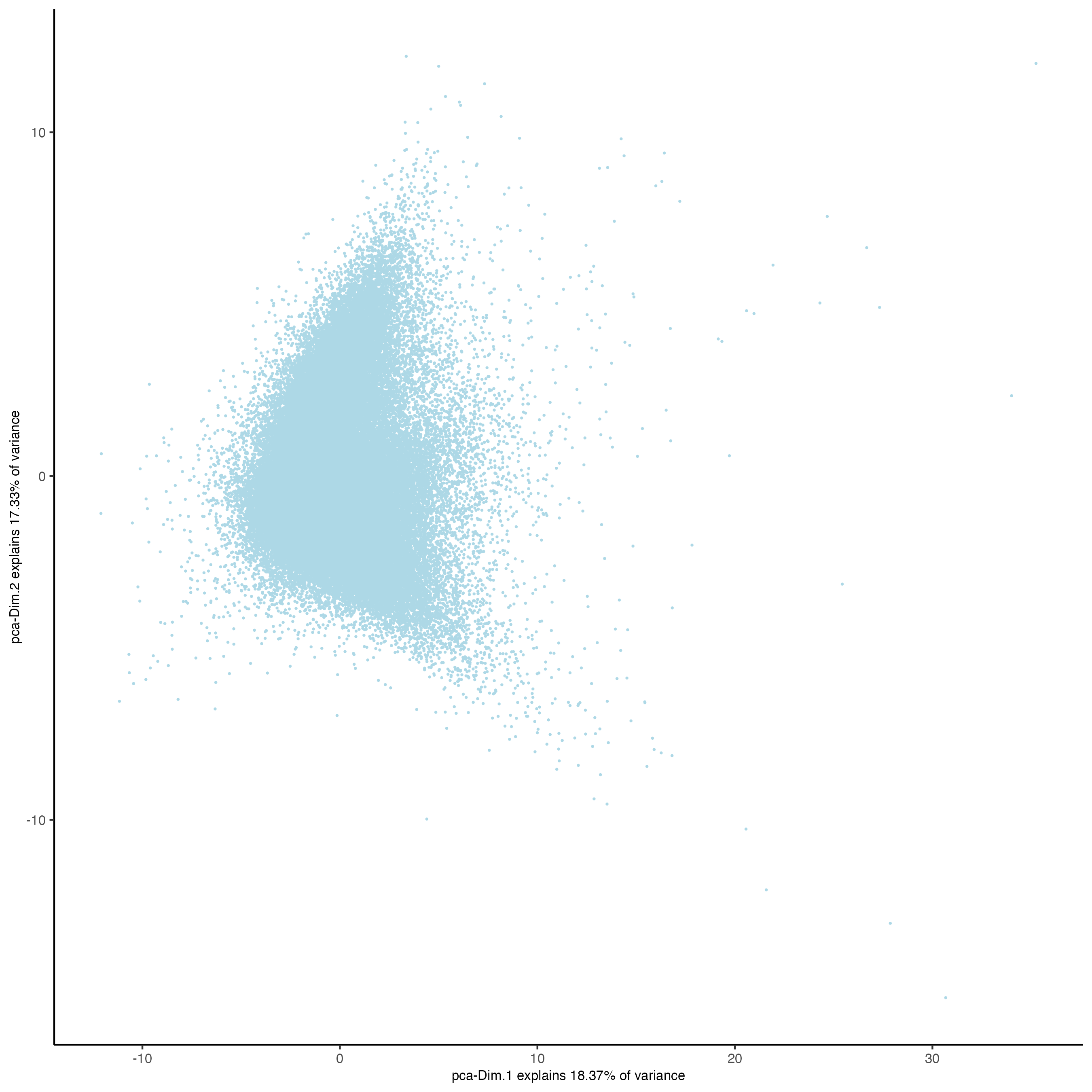
# UMAP
codex_test <- runUMAP(codex_tesTRUE,
dimensions_to_use = 1:14,
n_components = 2,
n_threads = 12)
plotUMAP(gobject = codex_tesTRUE,
point_shape = "no_border",
point_size = 0.2,
save_param = list(save_name = "3_c_UMAP"))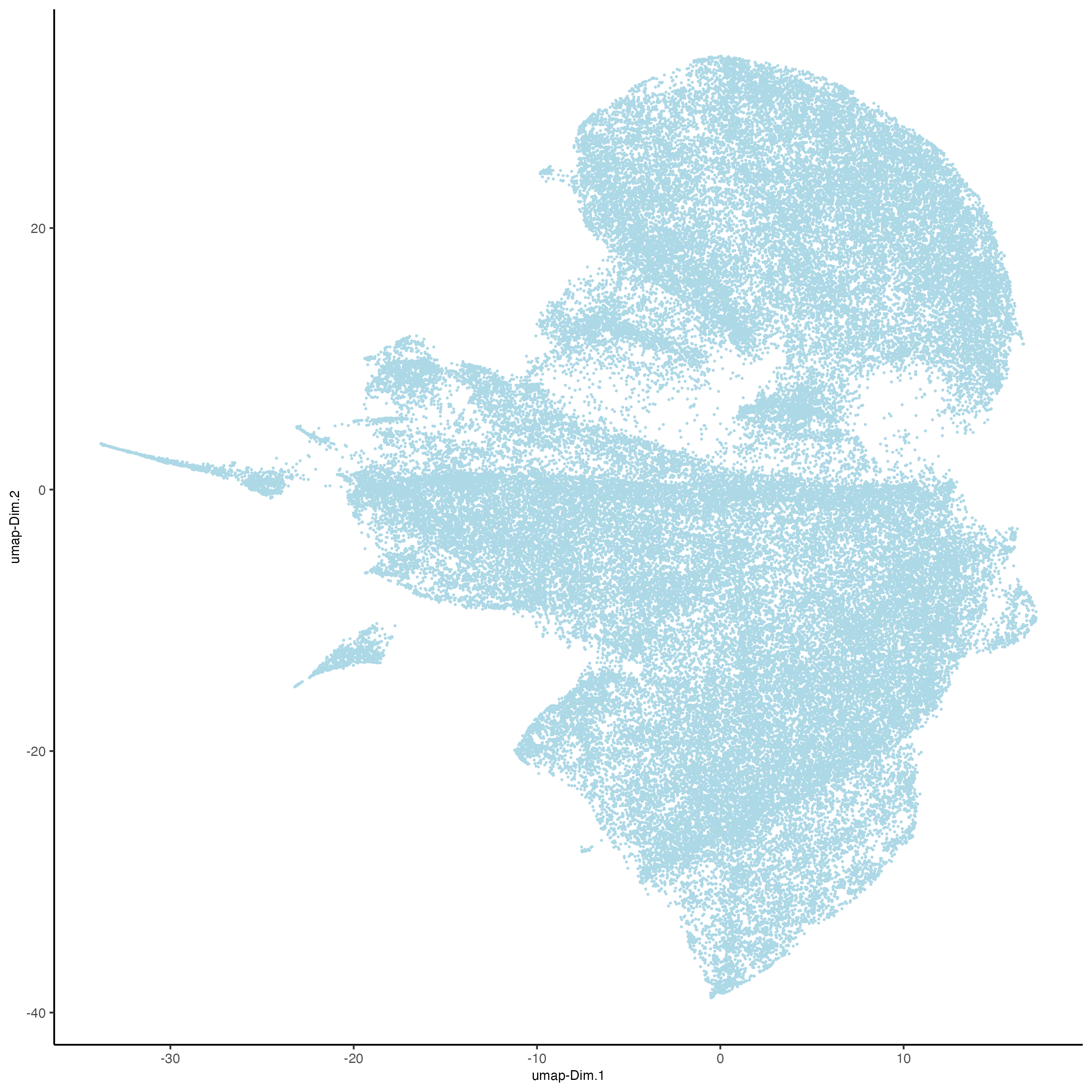
6 Cluster
## sNN network (default)
codex_test <- createNearestNetwork(gobject = codex_tesTRUE,
dimensions_to_use = 1:14,
k = 20)
## 0.1 resolution
codex_test <- doLeidenCluster(gobject = codex_tesTRUE,
resolution = 0.5,
n_iterations = 100,
name = "leiden")
codex_metadata <- pDataDT(codex_test)
leiden_colors <- getDistinctColors(length(unique(codex_metadata$leiden)))
names(leiden_colors) <- unique(codex_metadata$leiden)
plotUMAP(gobject = codex_tesTRUE,
cell_color = "leiden",
point_shape = "no_border",
point_size = 0.2,
cell_color_code = leiden_colors,
save_param = list(save_name = "4_a_UMAP"))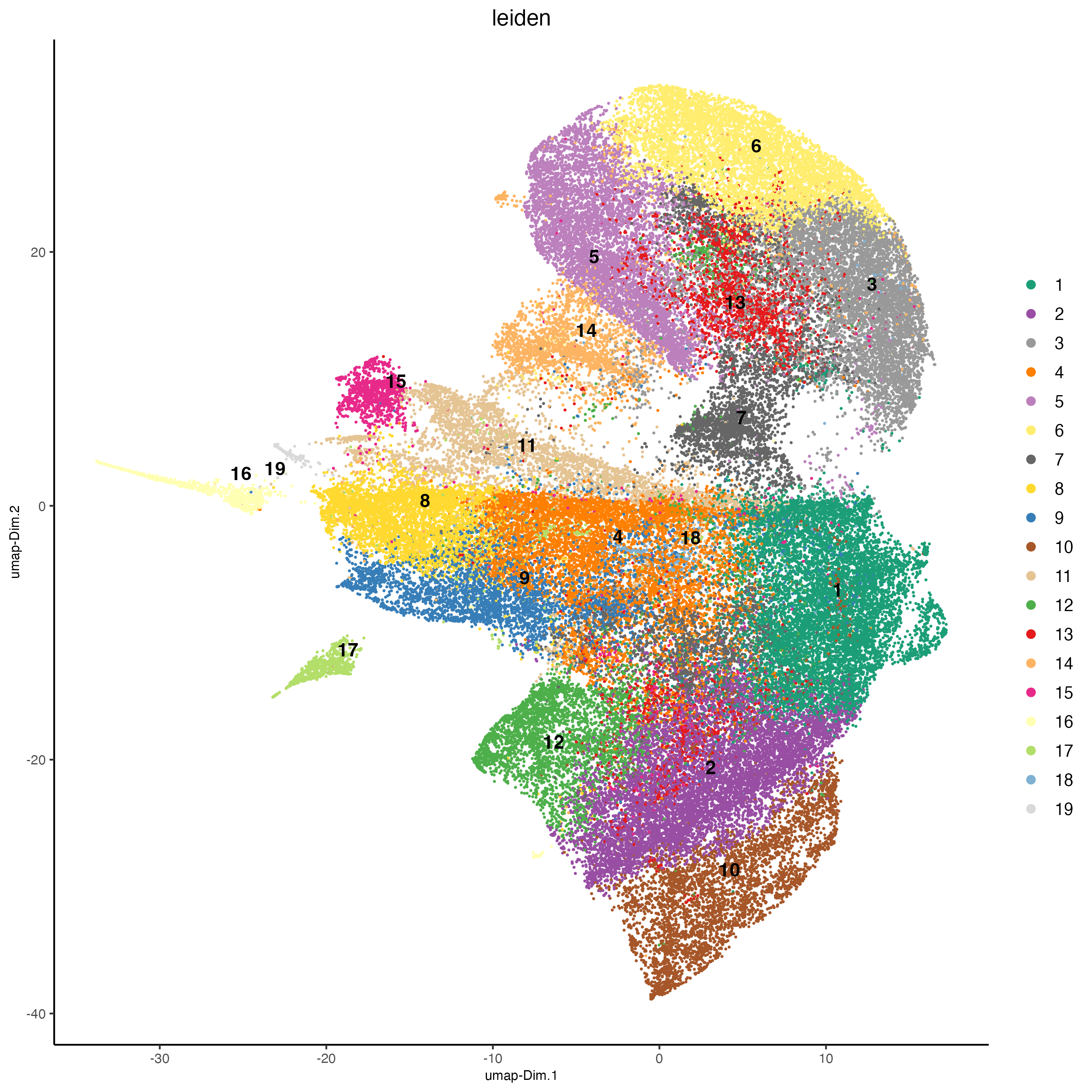
spatPlot(gobject = codex_tesTRUE,
cell_color = "leiden",
point_shape = "no_border",
point_size = 0.2,
cell_color_code = leiden_colors,
coord_fix_ratio = 1,
label_size = 2,
legend_text = 5,
legend_symbol_size = 2,
save_param = list(save_name = "4_b_spatplot"))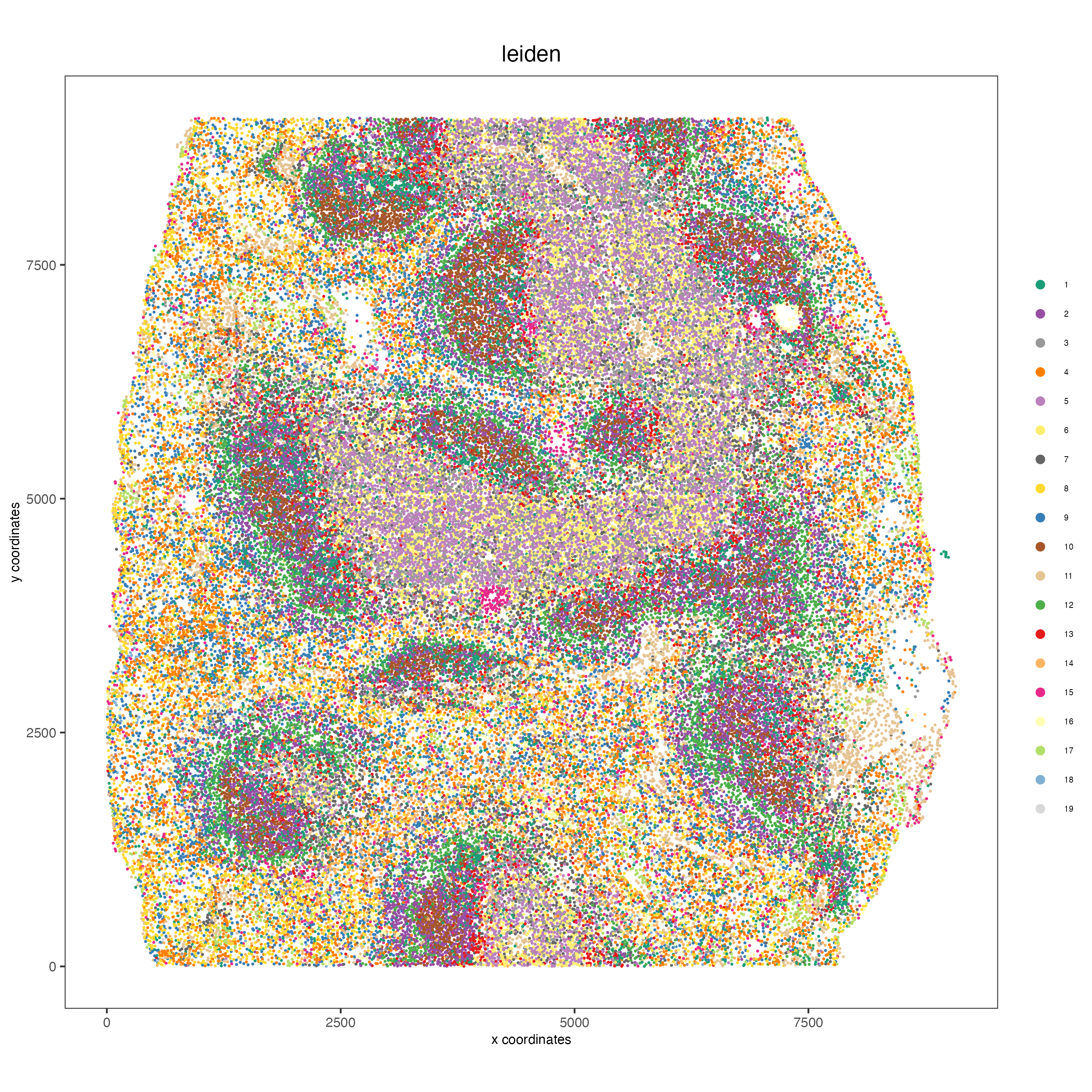
7 Co-visualize
spatDimPlot2D(gobject = codex_tesTRUE,
cell_color = "leiden",
spat_point_shape = "no_border",
spat_point_size = 0.2,
dim_point_shape = "no_border",
dim_point_size = 0.2,
cell_color_code = leiden_colors,
plot_alignment = "horizontal",
save_param = list(save_name = "5_a_spatdimplot"))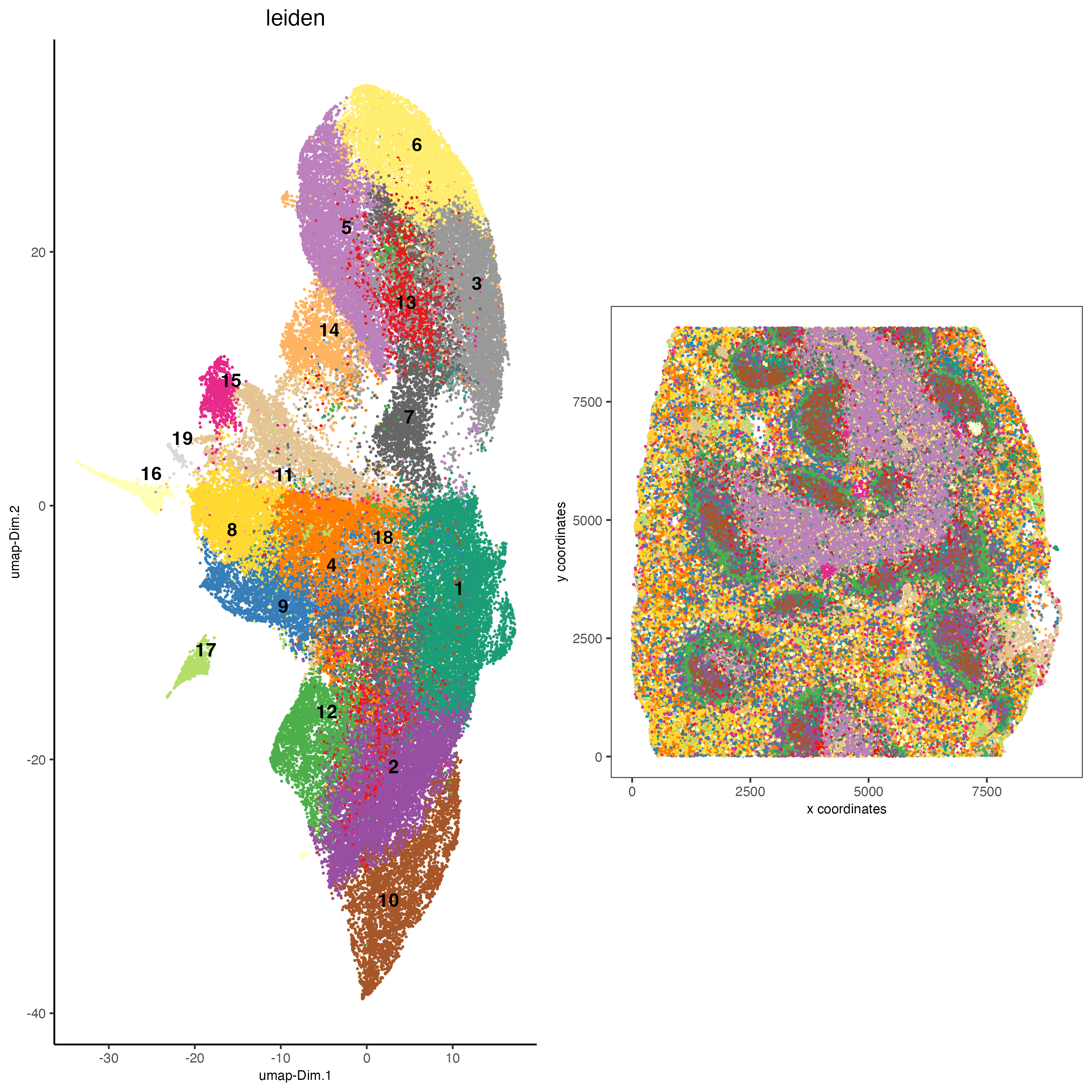
8 Differential expression
cluster_column <- "leiden"
markers_scran <- findMarkers_one_vs_all(gobject = codex_tesTRUE,
method = "scran",
expression_values = "normalized",
cluster_column = cluster_column,
min_feats = 3)
topgenes_scran <- unique(markers_scran[, head(.SD, 5), by = "cluster"][["feats"]])
plotMetaDataHeatmap(codex_tesTRUE,
expression_values = "normalized",
metadata_cols = cluster_column,
selected_feats = topgenes_scran,
y_text_size = 8,
show_values = "zscores_rescaled",
save_param = list(save_name = "6_a_metaheatmap"))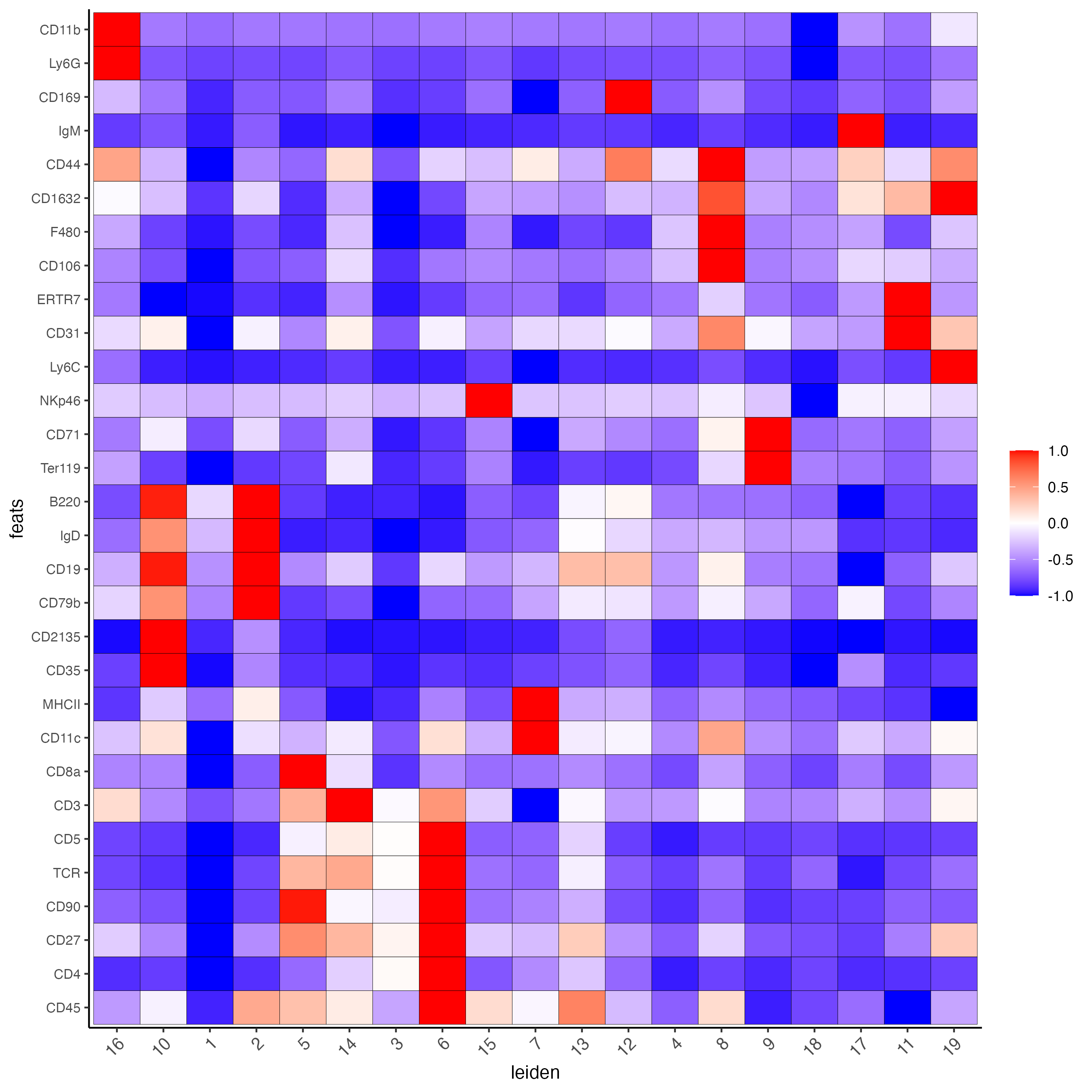
topgenes_scran <- markers_scran[, head(.SD, 1), by = "cluster"]$feats
violinPlot(codex_tesTRUE,
feats = unique(topgenes_scran)[1:8],
cluster_column = cluster_column,
strip_text = 8,
strip_position = "right",
save_param = list(save_name = "6_b_violinplot"))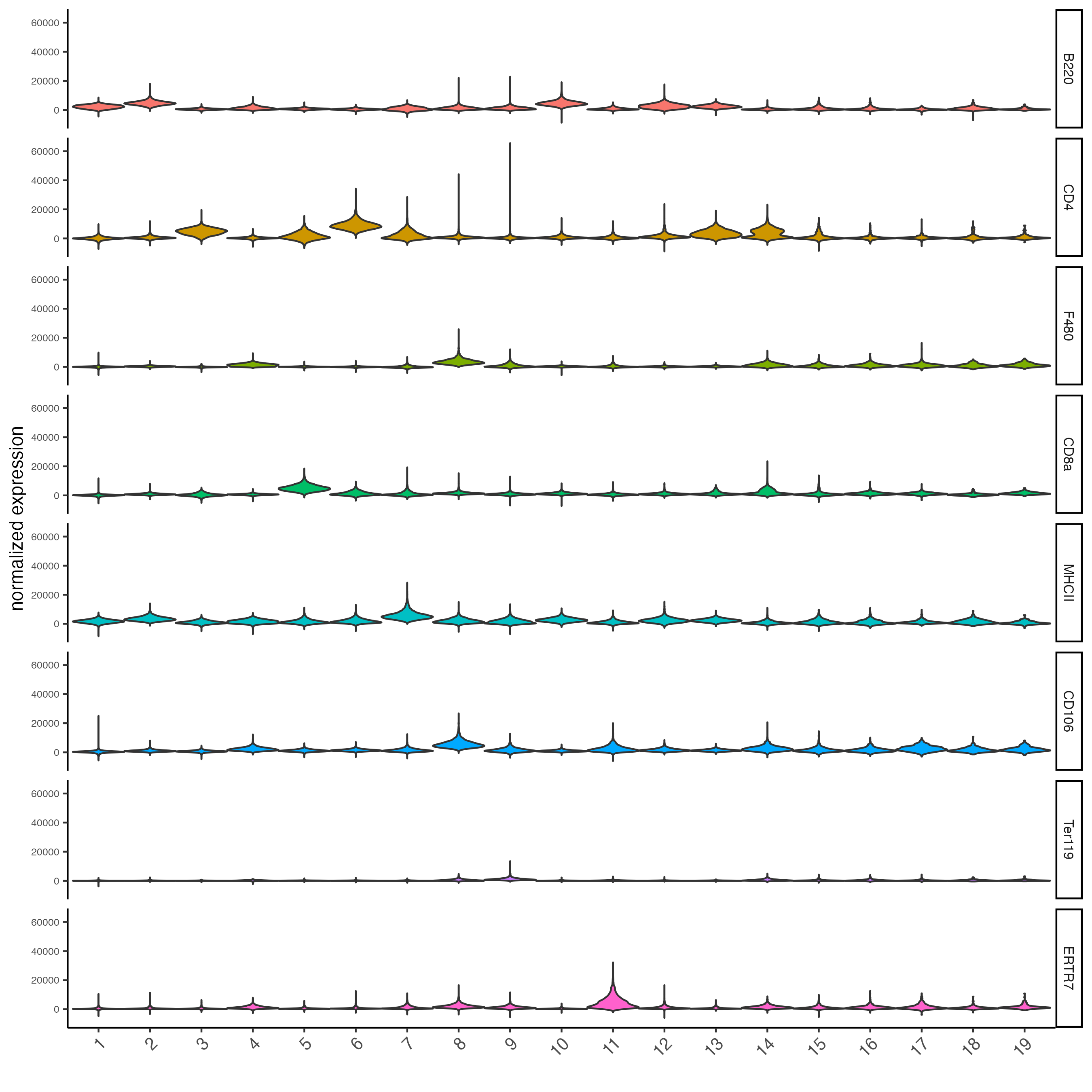
# gini
markers_gini <- findMarkers_one_vs_all(gobject = codex_tesTRUE,
method = "gini",
expression_values = "normalized",
cluster_column = cluster_column,
min_feats = 5)
topgenes_gini <- unique(markers_gini[, head(.SD, 5), by = "cluster"][["feats"]])
plotMetaDataHeatmap(codex_tesTRUE,
expression_values = "normalized",
metadata_cols = cluster_column,
selected_feats = topgenes_gini,
show_values = "zscores_rescaled",
save_param = list(save_name = "6_c_metaheatmap"))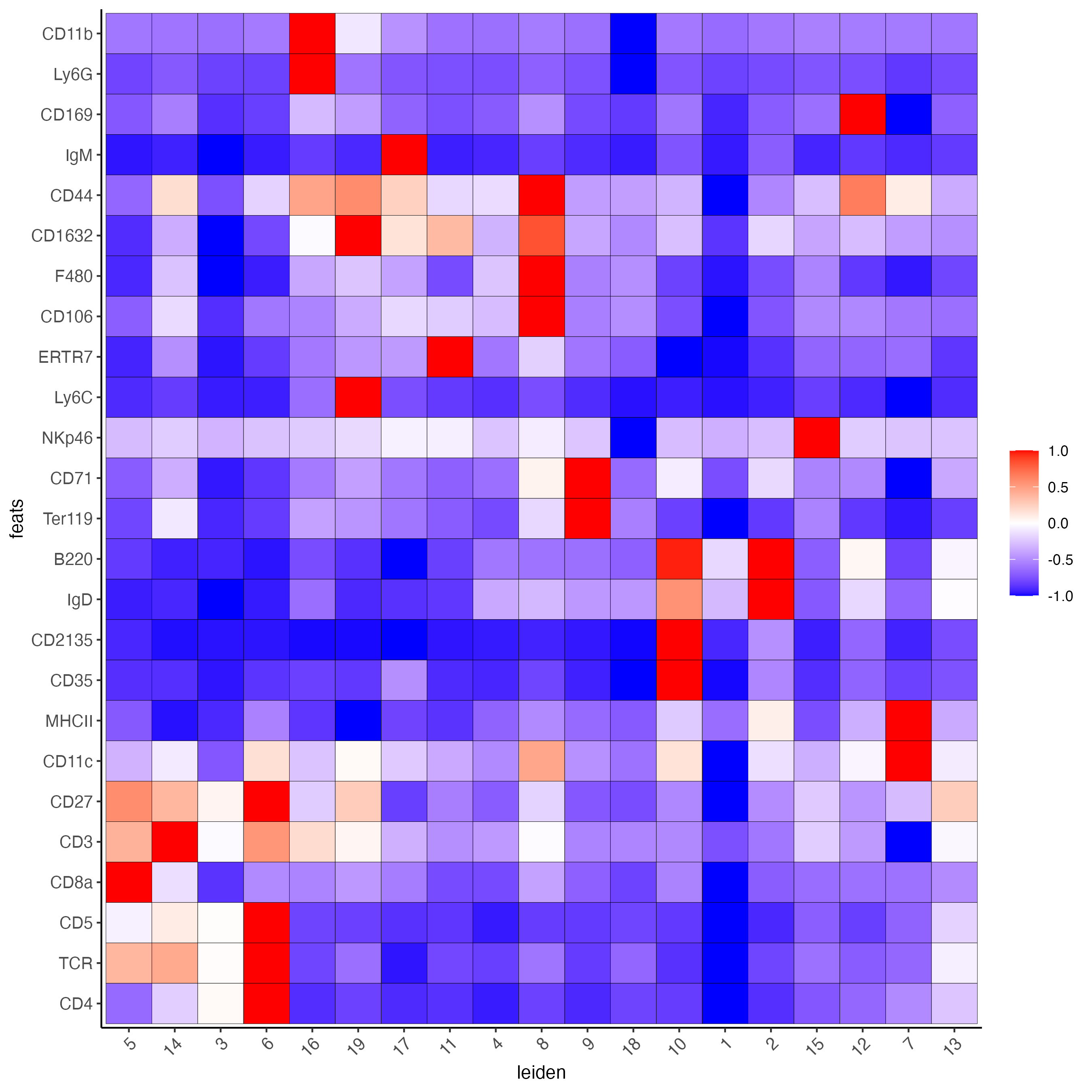
topgenes_gini <- markers_gini[, head(.SD, 1), by = "cluster"]$feats
violinPlot(codex_tesTRUE,
feats = unique(topgenes_gini),
cluster_column = cluster_column,
strip_text = 8,
strip_position = "right",
save_param = list(save_name = "6_d_violinplot"))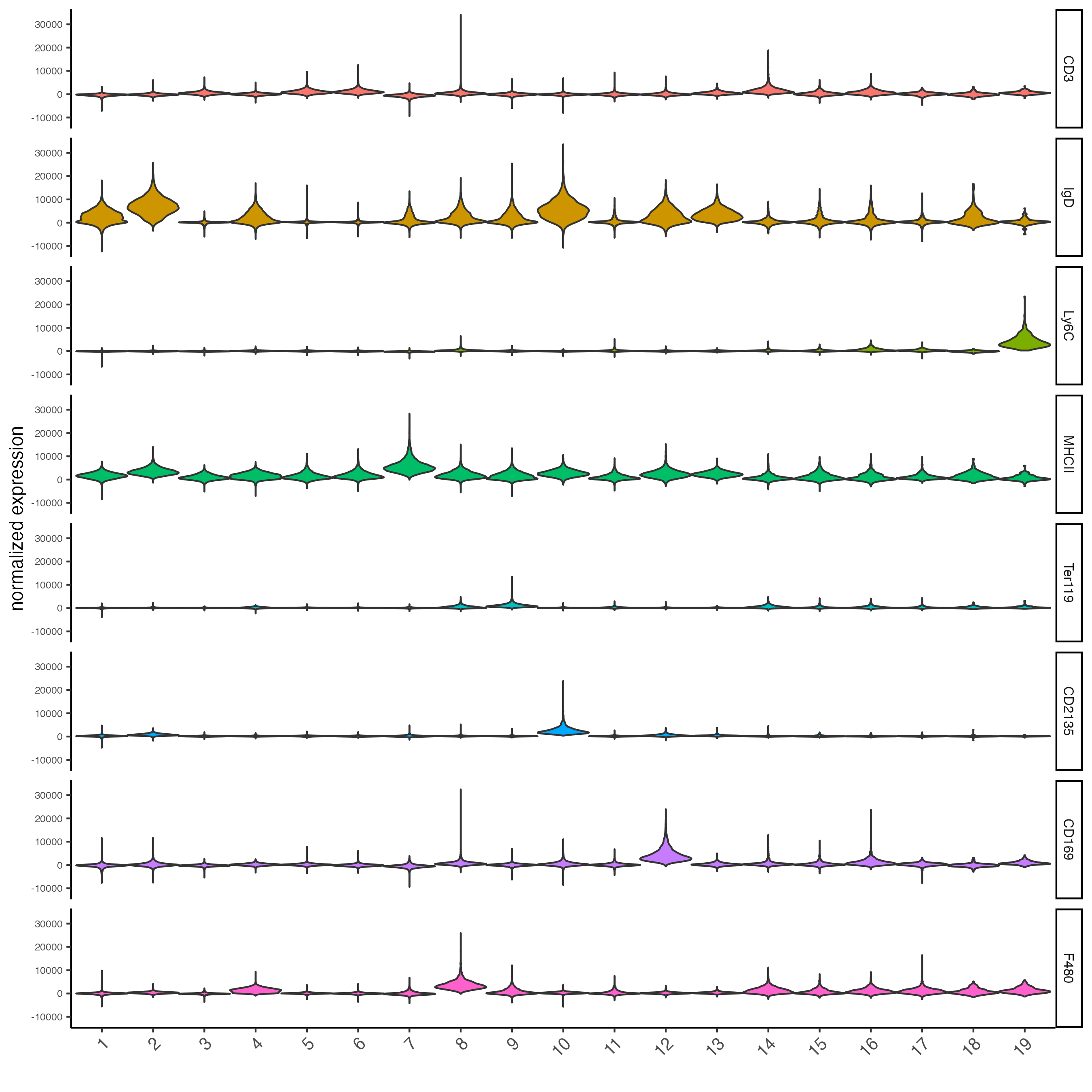
9 Cell type annotation
clusters_cell_types <- c("naive B cells", "B cells", "B cells", "naive B cells",
"B cells", "macrophages", "erythroblasts",
"erythroblasts", "erythroblasts", "CD8 + T cells",
"Naive T cells", "CD4+ T cells", "Naive T cells",
"CD4+ T cells", "Dendritic cells", "NK cells",
"Dendritic cells", "Plasma cells", "endothelial cells",
"monocytes")
names(clusters_cell_types) <- c(2, 15, 13, 5, 8, 9, 19, 1, 10, 3, 12, 14, 4, 6,
7, 16, 17, 18, 11, 20)
codex_test <- annotateGiotto(gobject = codex_tesTRUE,
annotation_vector = clusters_cell_types,
cluster_column = "leiden",
name = "cell_types")
plotUMAP(gobject = codex_tesTRUE,
cell_color = "cell_types",
point_shape = "no_border",
point_size = 0.2,
show_center_label = FALSE,
label_size = 2,
legend_text = 5,
legend_symbol_size = 2,
save_param = list(save_name = "7_a_umap_celltypes"))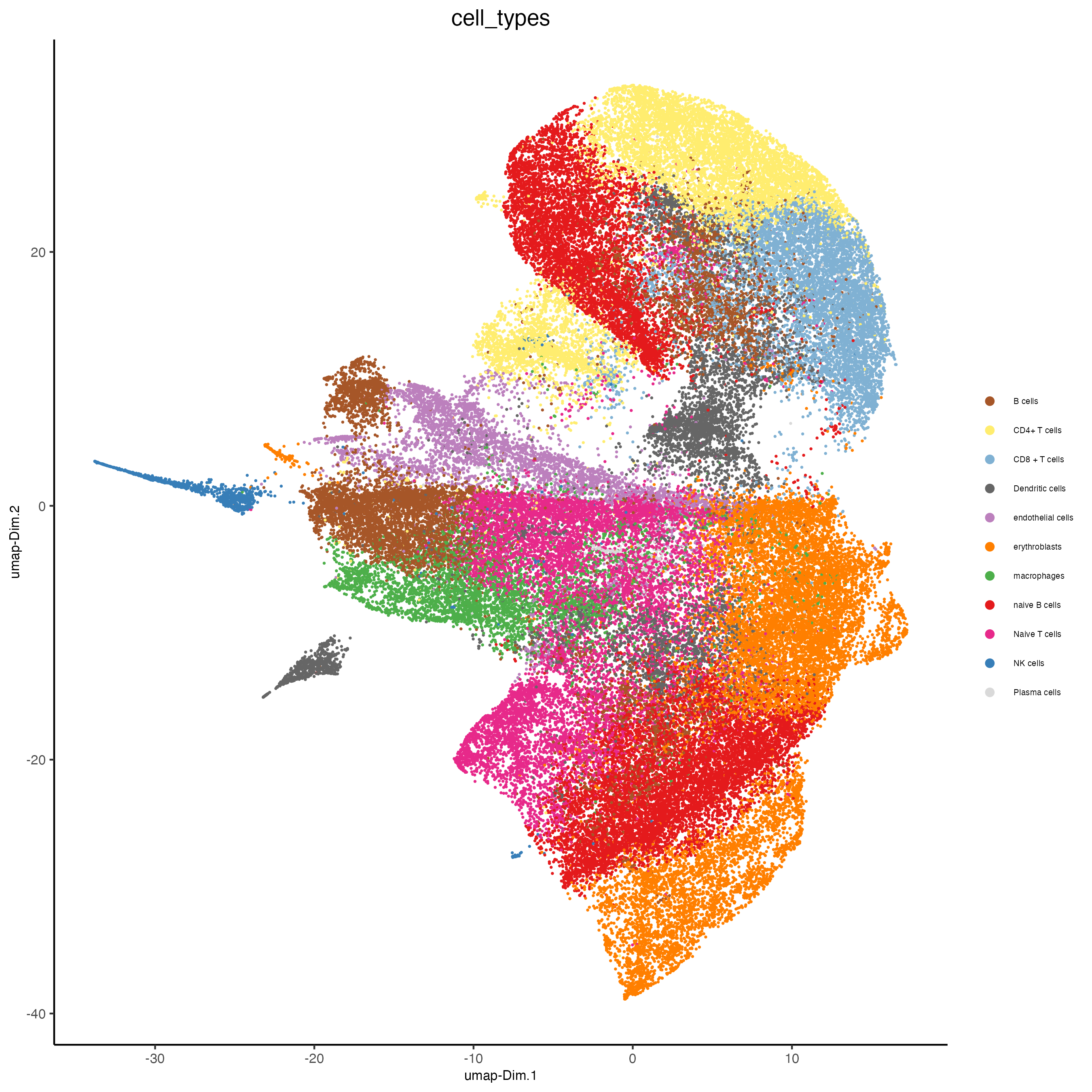
Or, this dataset comes with the imaging phenotype annotation
plotUMAP(gobject = codex_tesTRUE,
cell_color = "Imaging_phenotype_cell_type",
point_shape = "no_border",
point_size = 0.2,
show_center_label = FALSE,
label_size = 2,
legend_text = 5,
legend_symbol_size = 2,
save_param = list(save_name = "7_b_umap"))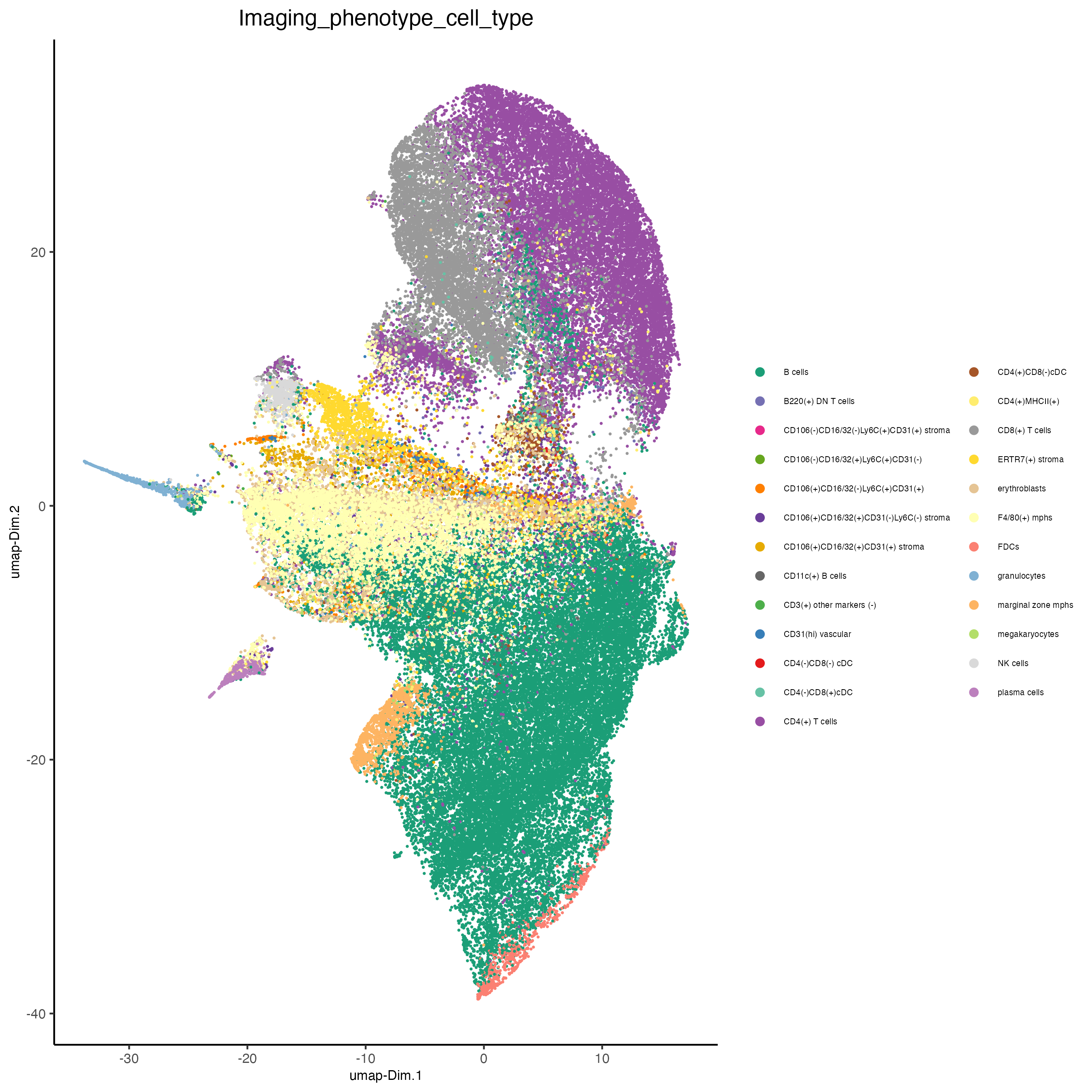
spatPlot(gobject = codex_tesTRUE,
cell_color = "Imaging_phenotype_cell_type",
point_shape = "no_border",
point_size = 0.2,
coord_fix_ratio = 1,
label_size = 2,
legend_text = 5,
legend_symbol_size = 2,
save_param = list(save_name = "7_c_spatplot"))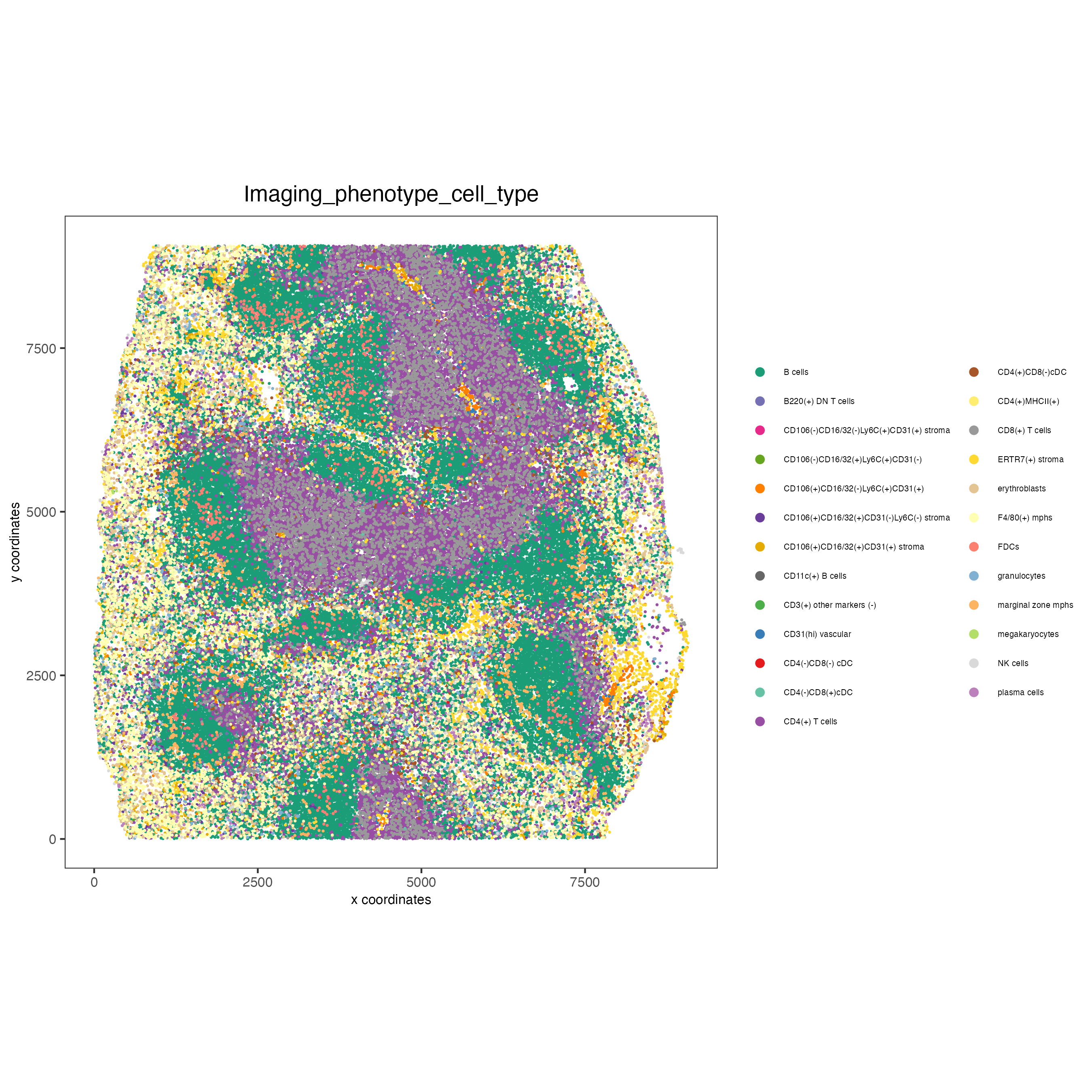
10 Visualize cell types and gene expression in selected zones
cell_metadatadata <- pDataDT(codex_test)
subset_cell_ids <- cell_metadatadata[sample_Xtile_Ytile=="BALBc-3_X04_Y08"]$cell_ID
codex_test_zone1 <- subsetGiotto(codex_tesTRUE,
cell_ids = subset_cell_ids)
plotUMAP(gobject = codex_test_zone1,
cell_color = "Imaging_phenotype_cell_type",
point_shape = "no_border",
point_size = 1,
show_center_label = FALSE,
label_size = 2,
legend_text = 5,
legend_symbol_size = 2,
save_param = list(save_name = "8_a_umap"))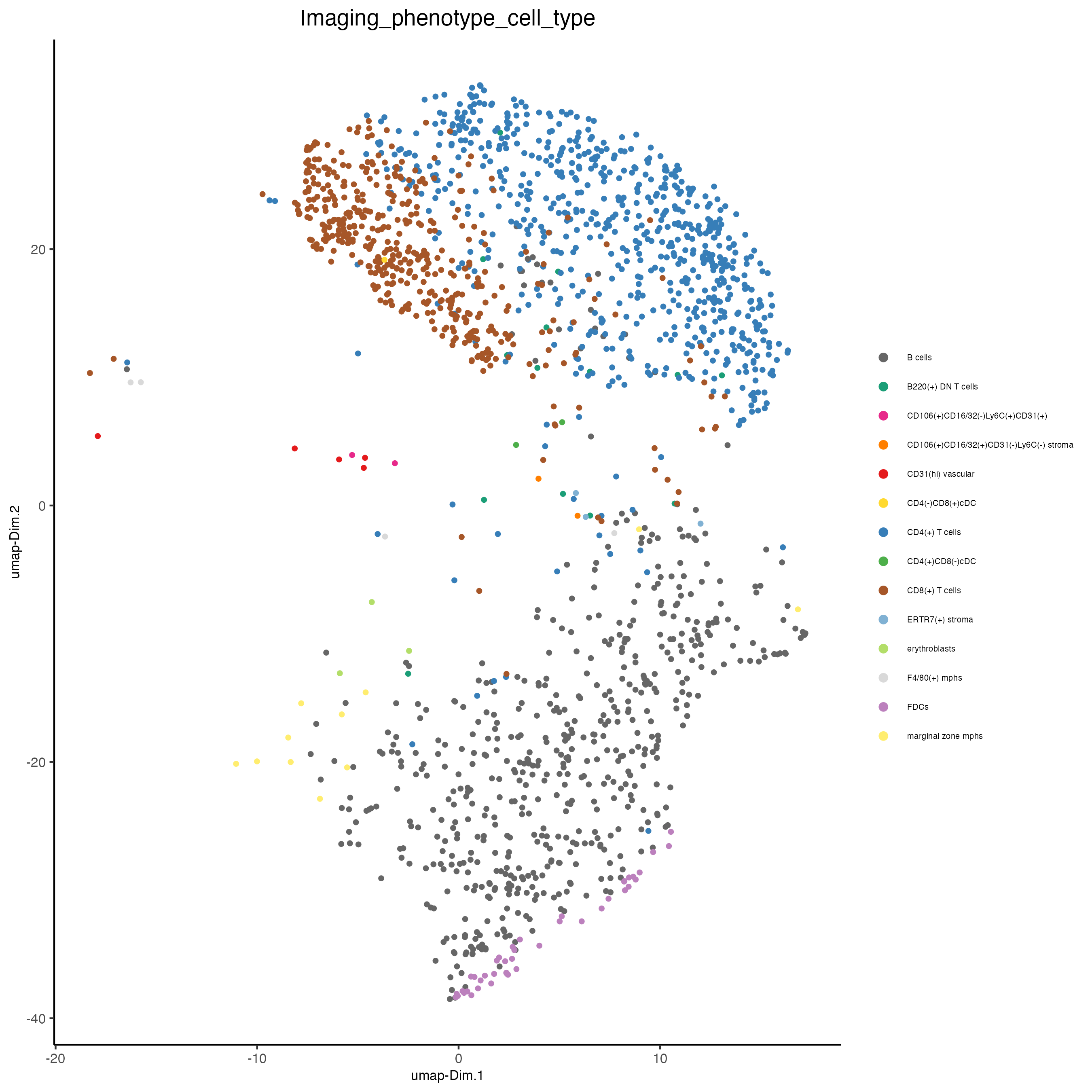
spatPlot(gobject = codex_test_zone1,
cell_color = "Imaging_phenotype_cell_type",
point_shape = "no_border",
point_size = 1,
coord_fix_ratio = 1,
label_size = 2,
legend_text = 5,
legend_symbol_size = 2,
save_param = list(save_name = "8_b_spatplot"))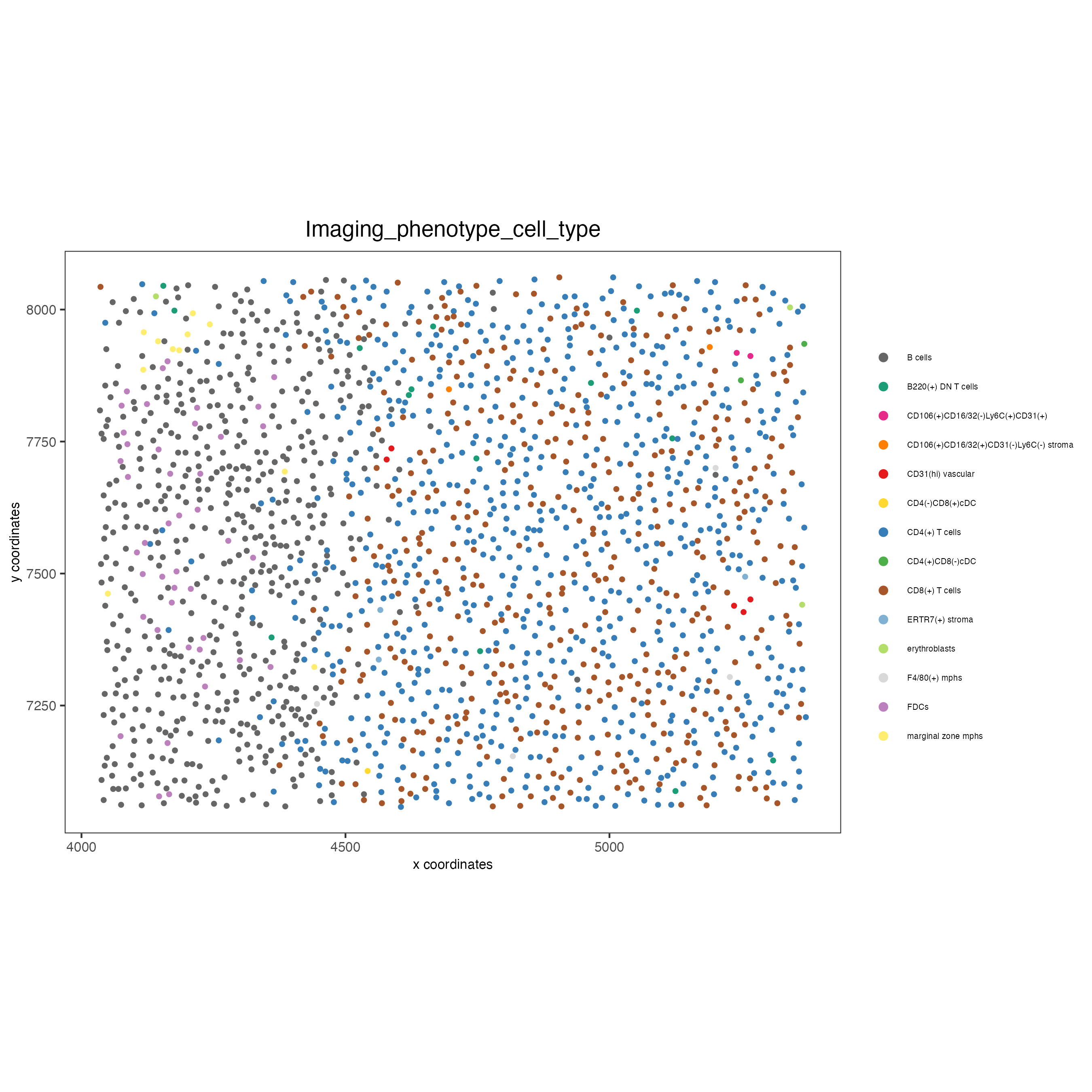
spatDimFeatPlot2D(codex_test_zone1,
expression_values = "scaled",
feats = c("CD8a","CD19"),
spat_point_shape = "no_border",
dim_point_shape = "no_border",
cell_color_gradient = c("darkblue", "white", "red"),
save_param = list(save_name = "8_c_spatdimplot"))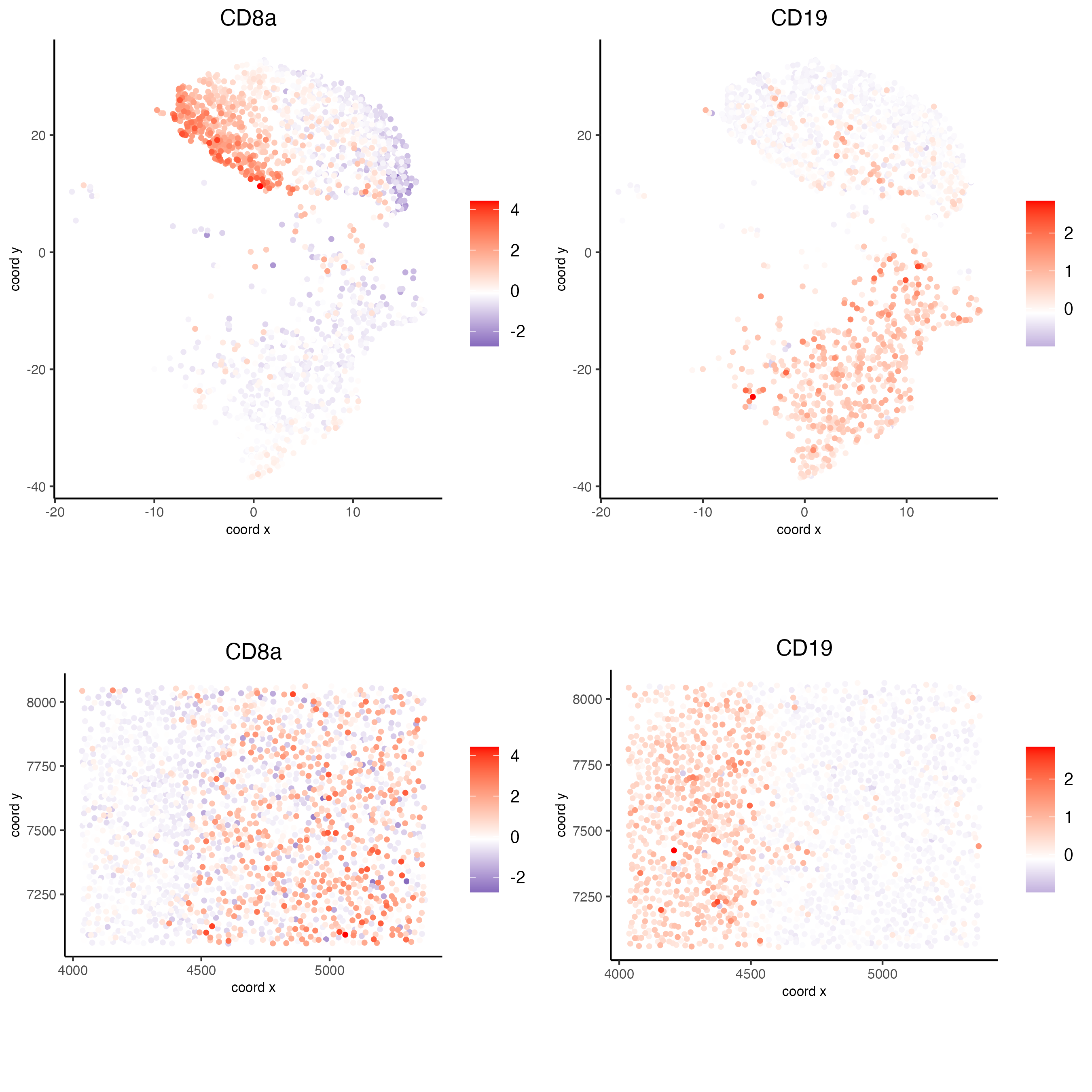
Test on another region:
cell_metadatadata <- pDataDT(codex_test)
subset_cell_ids <- cell_metadatadata[sample_Xtile_Ytile=="BALBc-3_X04_Y03"]$cell_ID
codex_test_zone2 <- subsetGiotto(codex_tesTRUE,
cell_ids = subset_cell_ids)
plotUMAP(gobject = codex_test_zone2,
cell_color = "Imaging_phenotype_cell_type",
point_shape = "no_border",
point_size = 1,
show_center_label = FALSE,
label_size = 2,
legend_text = 5,
legend_symbol_size = 2,
save_param = list(save_name = "8_d_umap"))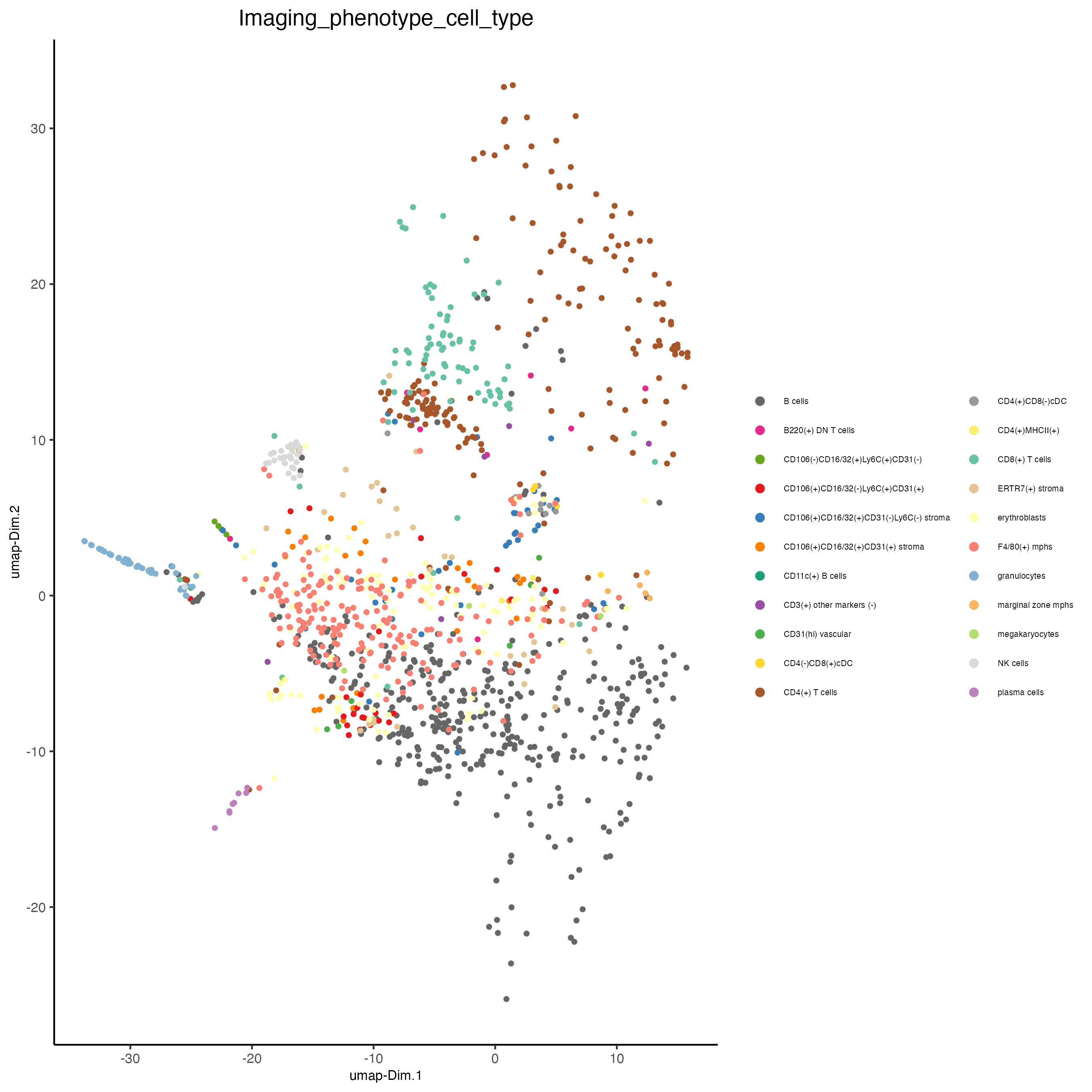
spatPlot(gobject = codex_test_zone2,
cell_color = "Imaging_phenotype_cell_type",
point_shape = "no_border",
point_size = 1,
coord_fix_ratio = 1,
label_size = 2,
legend_text = 5,
legend_symbol_size = 2,
save_param = list(save_name = "8_e_spatPlot"))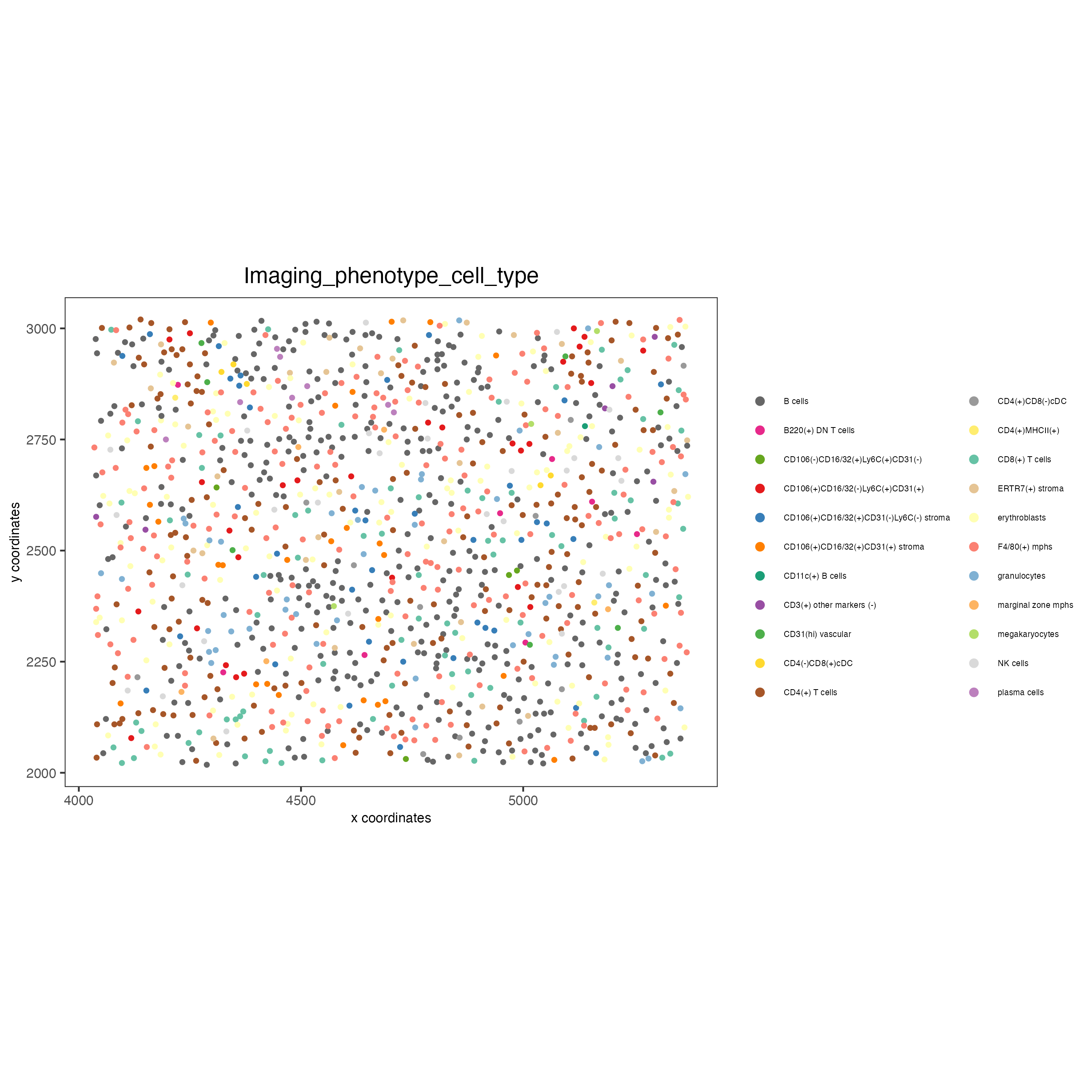
spatDimFeatPlot2D(codex_test_zone2,
expression_values = "scaled",
feats = c("CD4", "CD106"),
spat_point_shape = "no_border",
dim_point_shape = "no_border",
cell_color_gradient = c("darkblue", "white", "red"),
save_param = list(save_name = "8_f_spatdimgeneplot"))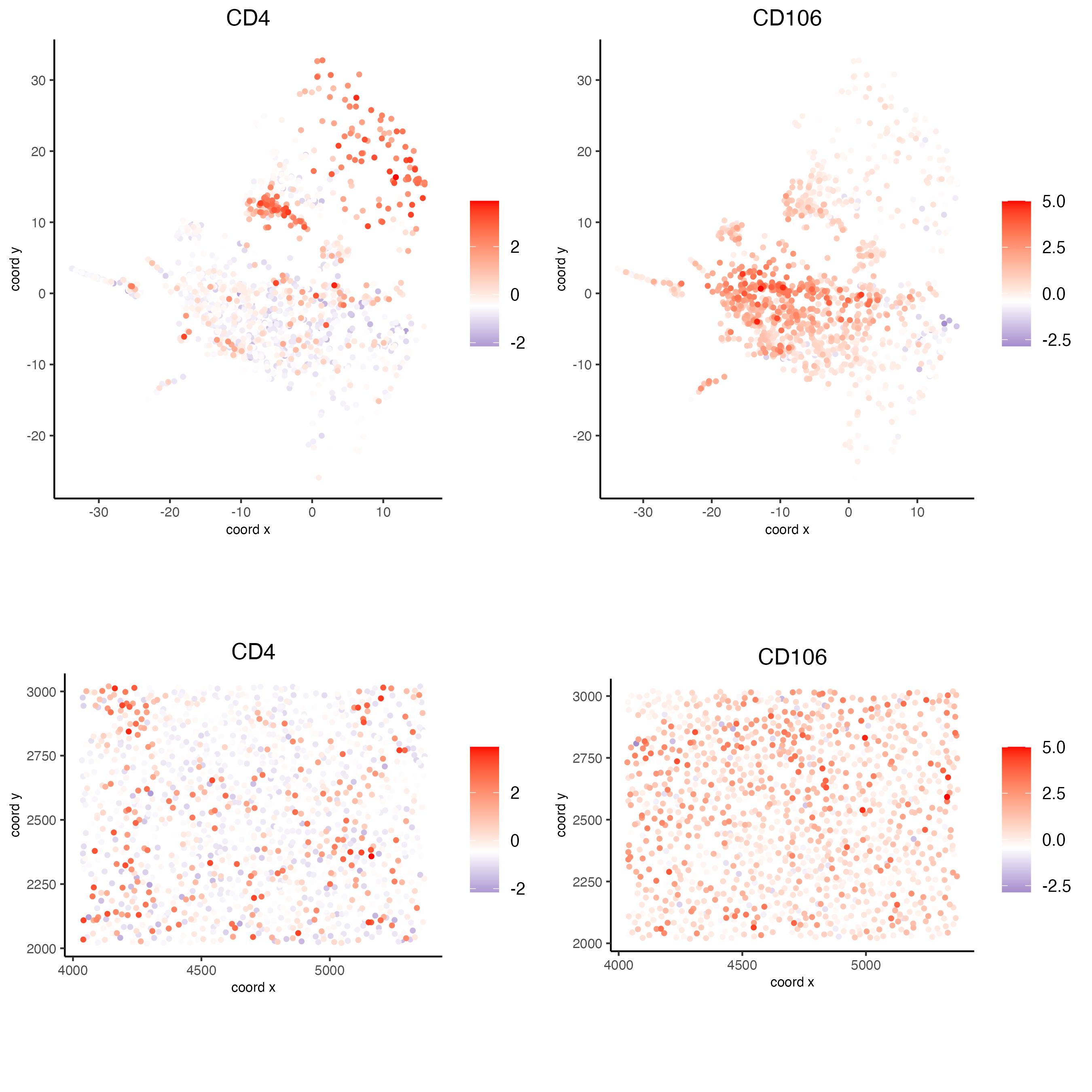
11 Session info
R version 4.3.2 (2023-10-31)
Platform: x86_64-apple-darwin20 (64-bit)
Running under: macOS Sonoma 14.3.1
Matrix products: default
BLAS: /System/Library/Frameworks/Accelerate.framework/Versions/A/Frameworks/vecLib.framework/Versions/A/libBLAS.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.3-x86_64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: America/New_York
tzcode source: internal
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] GiottoData_0.2.6.2 GiottoUtils_0.1.5 Giotto_4.0.2 GiottoClass_0.1.3
loaded via a namespace (and not attached):
[1] colorRamp2_0.1.0 bitops_1.0-7 rlang_1.1.3
[4] magrittr_2.0.3 RcppAnnoy_0.0.22 matrixStats_1.2.0
[7] compiler_4.3.2 DelayedMatrixStats_1.24.0 png_0.1-8
[10] systemfonts_1.0.5 vctrs_0.6.5 pkgconfig_2.0.3
[13] SpatialExperiment_1.12.0 crayon_1.5.2 fastmap_1.1.1
[16] backports_1.4.1 magick_2.8.2 XVector_0.42.0
[19] scuttle_1.12.0 labeling_0.4.3 utf8_1.2.4
[22] rmarkdown_2.25 ragg_1.2.7 bluster_1.12.0
[25] xfun_0.42 beachmat_2.18.0 zlibbioc_1.48.0
[28] GenomeInfoDb_1.38.6 jsonlite_1.8.8 flashClust_1.01-2
[31] pak_0.7.1 DelayedArray_0.28.0 BiocParallel_1.36.0
[34] terra_1.7-71 irlba_2.3.5.1 parallel_4.3.2
[37] cluster_2.1.6 R6_2.5.1 RColorBrewer_1.1-3
[40] limma_3.58.1 reticulate_1.35.0 GenomicRanges_1.54.1
[43] estimability_1.4.1 Rcpp_1.0.12 SummarizedExperiment_1.32.0
[46] knitr_1.45 R.utils_2.12.3 IRanges_2.36.0
[49] igraph_2.0.1.1 Matrix_1.6-5 tidyselect_1.2.0
[52] rstudioapi_0.15.0 abind_1.4-5 yaml_2.3.8
[55] codetools_0.2-19 lattice_0.22-5 tibble_3.2.1
[58] Biobase_2.62.0 withr_3.0.0 evaluate_0.23
[61] pillar_1.9.0 MatrixGenerics_1.14.0 checkmate_2.3.1
[64] DT_0.31 stats4_4.3.2 dbscan_1.1-12
[67] generics_0.1.3 RCurl_1.98-1.14 S4Vectors_0.40.2
[70] ggplot2_3.4.4 sparseMatrixStats_1.14.0 munsell_0.5.0
[73] scales_1.3.0 gtools_3.9.5 xtable_1.8-4
[76] leaps_3.1 glue_1.7.0 metapod_1.10.1
[79] emmeans_1.10.0 scatterplot3d_0.3-44 tools_4.3.2
[82] GiottoVisuals_0.1.4 BiocNeighbors_1.20.2 data.table_1.15.0
[85] ScaledMatrix_1.10.0 locfit_1.5-9.8 scran_1.30.2
[88] mvtnorm_1.2-4 cowplot_1.1.3 grid_4.3.2
[91] edgeR_4.0.14 colorspace_2.1-0 SingleCellExperiment_1.24.0
[94] GenomeInfoDbData_1.2.11 BiocSingular_1.18.0 rsvd_1.0.5
[97] cli_3.6.2 textshaping_0.3.7 fansi_1.0.6
[100] S4Arrays_1.2.0 dplyr_1.1.4 uwot_0.1.16
[103] gtable_0.3.4 R.methodsS3_1.8.2 digest_0.6.34
[106] progressr_0.14.0 BiocGenerics_0.48.1 dqrng_0.3.2
[109] SparseArray_1.2.3 ggrepel_0.9.5 FactoMineR_2.9
[112] rjson_0.2.21 htmlwidgets_1.6.4 farver_2.1.1
[115] htmltools_0.5.7 R.oo_1.26.0 lifecycle_1.0.4
[118] multcompView_0.1-9 statmod_1.5.0 MASS_7.3-60.0.1