Hidden Markov Random Field (HMRF) models capture spatial dependencies and segment tissue regions based on shared and gene expression patterns.
1 Setup and load example dataset
# Ensure Giotto Suite is installed
if(!"Giotto" %in% installed.packages()) {
pak::pkg_install("drieslab/Giotto")
}
# Ensure Giotto Data is installed
if(!"GiottoData" %in% installed.packages()) {
pak::pkg_install("drieslab/GiottoData")
}
library(Giotto)
# Ensure the Python environment for Giotto has been installed
genv_exists <- checkGiottoEnvironment()
if(!genv_exists){
# The following command need only be run once to install the Giotto environment
installGiottoEnvironment()
}
# load the object
g <- GiottoData::loadGiottoMini("visium")2 Run HMRF
Get the top genes per spatial co-expression module. For more information about how to calculate these genes, see the tutorial for Detection of spatial co-expression modules.
spatial_genes <- c("Hlf", "Amotl1", "Adarb1", "Rab3c",
"Prkcd", "Prph", "Tcf7l2", "Bok",
"Ptpn4", "Rgs16", "Cnp", "Bcas1",
"Qdpr", "Plekhb1", "Cryab", "Mobp",
"Stx1a", "Arpp19", "Ttc9b", "Fam163b",
"Igfbp6", "Ngef", "Lamp5", "Dkk3",
"Dkkl1", "Tbr1", "Cabp7", "Gria1",
"Cpne6", "Bhlhe22", "Lct", "1700001C02Rik",
"Crym", "Wfs1", "Scn3b", "Spink8",
"Syn2", "Nptxr", "Ppp1r1a", "Kcnip2",
"Nptx1", "Cplx2", "Snca", "2010300C02Rik",
"Itpka", "Hpca")Do HMRF with different betas the top genes per spatial co-expression module.
In a real-size dataset, this step may take several minutes to run.
4 Plot the spatial distribution of the HMRF domains.
spatPlot2D(gobject = g,
cell_color = "HMRF_k20_b.0")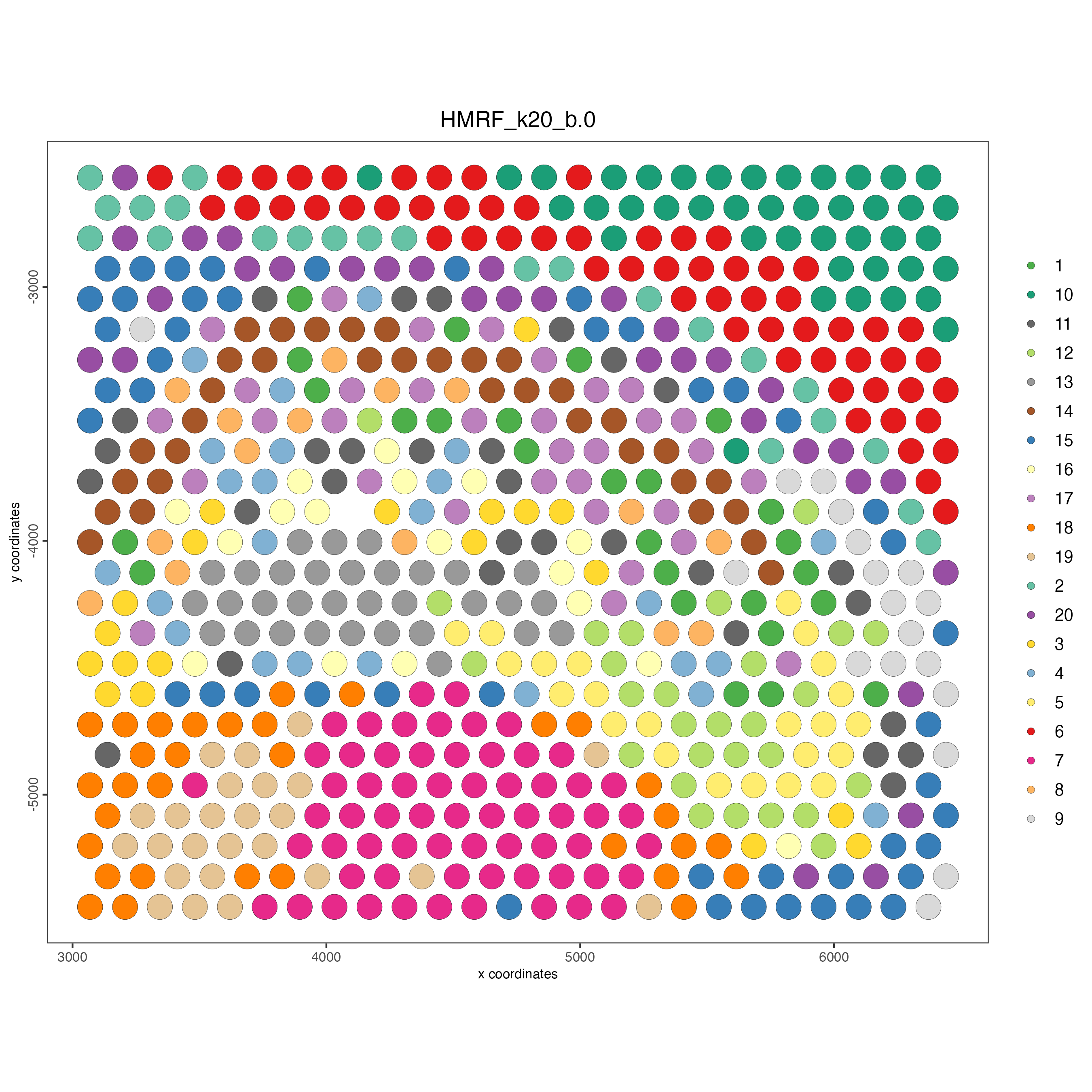
5 Session Info
R version 4.4.1 (2024-06-14)
Platform: x86_64-apple-darwin20
Running under: macOS 15.0
Matrix products: default
BLAS: /System/Library/Frameworks/Accelerate.framework/Versions/A/Frameworks/vecLib.framework/Versions/A/libBLAS.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.4-x86_64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: America/New_York
tzcode source: internal
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] Giotto_4.1.3 GiottoClass_0.4.0
loaded via a namespace (and not attached):
[1] colorRamp2_0.1.0 rlang_1.1.4
[3] magrittr_2.0.3 GiottoUtils_0.2.0
[5] matrixStats_1.4.1 compiler_4.4.1
[7] systemfonts_1.1.0 png_0.1-8
[9] callr_3.7.6 vctrs_0.6.5
[11] pkgconfig_2.0.3 SpatialExperiment_1.14.0
[13] crayon_1.5.3 fastmap_1.2.0
[15] backports_1.5.0 magick_2.8.5
[17] XVector_0.44.0 labeling_0.4.3
[19] utf8_1.2.4 rmarkdown_2.28
[21] tzdb_0.4.0 UCSC.utils_1.0.0
[23] ps_1.8.0 ragg_1.3.3
[25] purrr_1.0.2 xfun_0.47
[27] zlibbioc_1.50.0 GenomeInfoDb_1.40.1
[29] jsonlite_1.8.9 DelayedArray_0.30.1
[31] terra_1.7-78 parallel_4.4.1
[33] R6_2.5.1 RColorBrewer_1.1-3
[35] reticulate_1.39.0 GenomicRanges_1.56.1
[37] datapasta_3.1.0 scattermore_1.2
[39] Rcpp_1.0.13 SummarizedExperiment_1.34.0
[41] knitr_1.48 R.utils_2.12.3
[43] readr_2.1.5 IRanges_2.38.1
[45] Matrix_1.7-0 igraph_2.0.3
[47] tidyselect_1.2.1 rstudioapi_0.16.0
[49] abind_1.4-8 yaml_2.3.10
[51] codetools_0.2-20 processx_3.8.4
[53] lattice_0.22-6 tibble_3.2.1
[55] Biobase_2.64.0 withr_3.0.1
[57] evaluate_1.0.0 desc_1.4.3
[59] pillar_1.9.0 MatrixGenerics_1.16.0
[61] checkmate_2.3.2 stats4_4.4.1
[63] plotly_4.10.4 generics_0.1.3
[65] dbscan_1.2-0 hms_1.1.3
[67] S4Vectors_0.42.1 ggplot2_3.5.1
[69] munsell_0.5.1 scales_1.3.0
[71] GiottoData_0.2.15 gtools_3.9.5
[73] glue_1.8.0 clipr_0.8.0
[75] lazyeval_0.2.2 tools_4.4.1
[77] GiottoVisuals_0.2.5 data.table_1.16.0
[79] fs_1.6.4 cowplot_1.1.3
[81] grid_4.4.1 tidyr_1.3.1
[83] colorspace_2.1-1 SingleCellExperiment_1.26.0
[85] GenomeInfoDbData_1.2.12 cli_3.6.3
[87] textshaping_0.4.0 fansi_1.0.6
[89] S4Arrays_1.4.1 viridisLite_0.4.2
[91] dplyr_1.1.4 gtable_0.3.5
[93] R.methodsS3_1.8.2 digest_0.6.37
[95] BiocGenerics_0.50.0 SparseArray_1.4.8
[97] ggrepel_0.9.6 rjson_0.2.23
[99] htmlwidgets_1.6.4 farver_2.1.2
[101] htmltools_0.5.8.1 pkgdown_2.1.1
[103] R.oo_1.26.0 lifecycle_1.0.4
[105] httr_1.4.7