1 Start Giotto
# Ensure Giotto Suite is installed.
if(!"Giotto" %in% installed.packages()) {
pak::pkg_install("drieslab/Giotto")
}
# Ensure GiottoData, a small, helper module for tutorials, is installed.
if(!"GiottoData" %in% installed.packages()) {
pak::pkg_install("drieslab/GiottoData")
}
# Ensure the Python environment for Giotto has been installed.
genv_exists <- Giotto::checkGiottoEnvironment()
if(!genv_exists){
# The following command need only be run once to install the Giotto environment.
Giotto::installGiottoEnvironment()
}2 Load the Giotto object
A small seqFISH data is available through the giottoData package.
# download data
seqfish_mini <- loadGiottoMini("seqfish",
python_path = NULL)2.1 Set Giotto instructions (optional)
How to work with Giotto instructions that are part of your Giotto object:
- Show the instructions associated with your Giotto object with showGiottoInstructions()
- Change one or more instructions with changeGiottoInstructions()
- Replace all instructions at once with replaceGiottoInstructions()
- Read or get a specific Giotto instruction with readGiottoInstructions()
# show instructions associated with giotto object (seqfish_mini)
showGiottoInstructions(seqfish_mini)
# Change one or more instructions
# to automatically save figures in save_dir, set save_plot to TRUE
results_folder <- "/path/to/results/"
seqfish_mini <- changeGiottoInstructions(seqfish_mini,
params = c("save_dir", "save_plot", "show_plot"),
new_values = c(results_folder,TRUE, TRUE))3 Processing steps
- Filter genes and cells based on detection frequencies.
- Normalize expression matrix (log transformation, scaling factor and/or z-scores)
- Add cell and gene statistics (optional)
- Adjust expression matrix for technical covariates or batches (optional). These results will be stored in the custom slot.
seqfish_mini <- filterGiotto(gobject = seqfish_mini,
expression_threshold = 0.5,
feat_det_in_min_cells = 20,
min_det_feats_per_cell = 0)
seqfish_mini <- normalizeGiotto(gobject = seqfish_mini,
scalefactor = 6000,
verbose = TRUE)
seqfish_mini <- addStatistics(gobject = seqfish_mini)
seqfish_mini <- adjustGiottoMatrix(gobject = seqfish_mini,
expression_values = "normalized",
covariate_columns = c("nr_feats", "total_expr"))4 Dimension reduction
- Identify highly variable features (HVF)
seqfish_mini <- calculateHVF(gobject = seqfish_mini)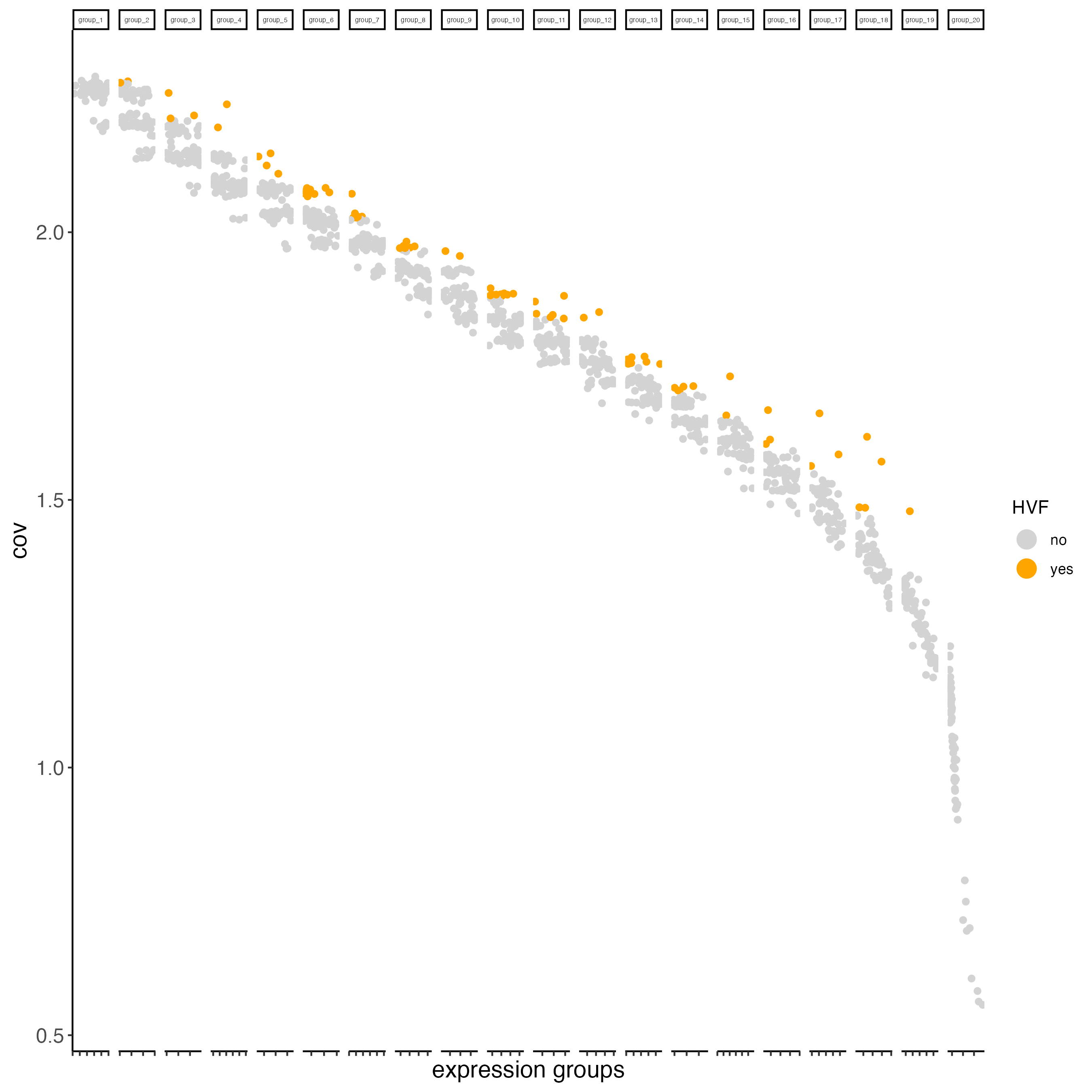
- Perform PCA
- Identify number of significant principal components (PCs)
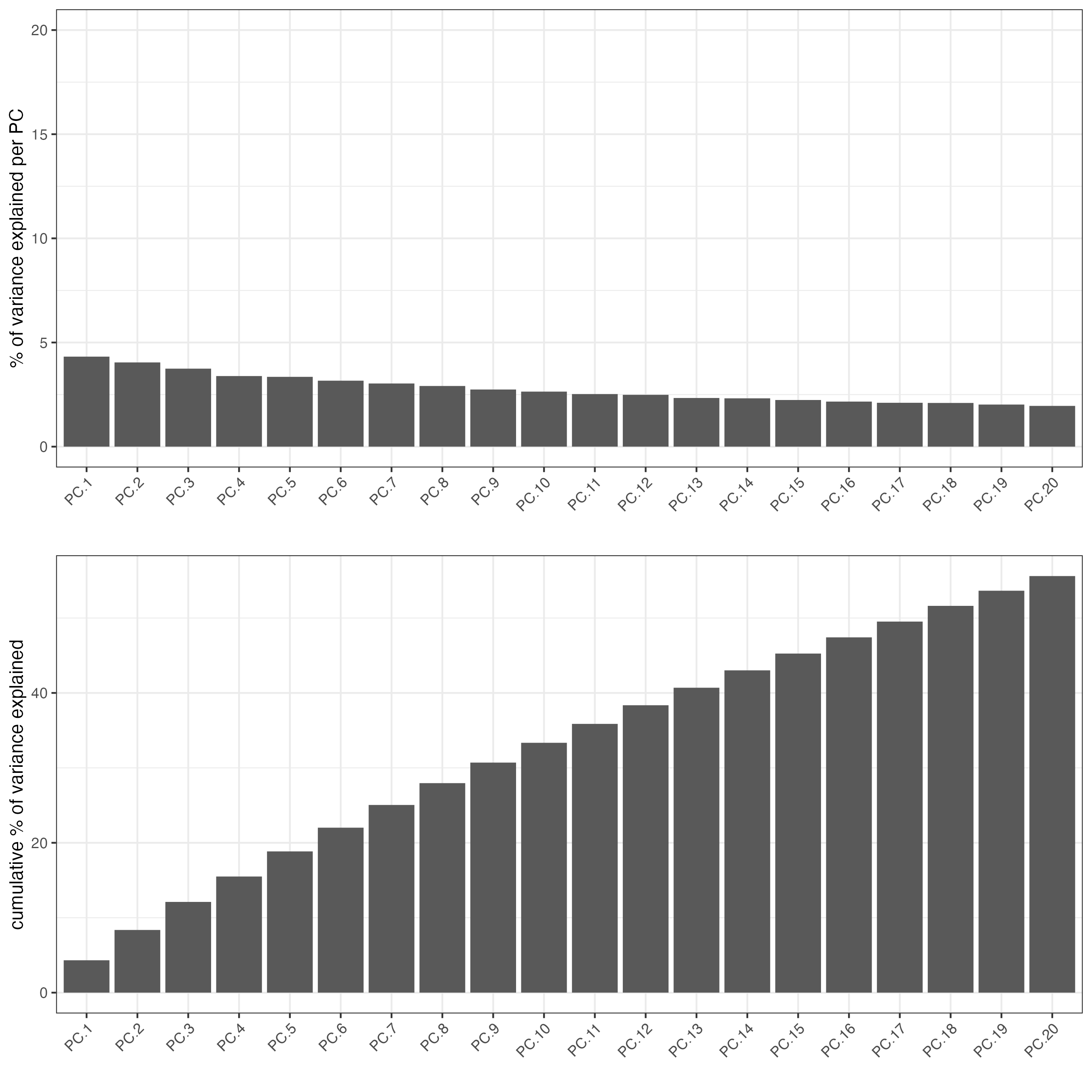
plotPCA(seqfish_mini)
- Run UMAP and/or t-SNE on PCs (or directly on matrix)
seqfish_mini <- runUMAP(seqfish_mini,
dimensions_to_use = 1:5,
n_threads = 2)
plotUMAP(gobject = seqfish_mini)
5 Clustering
- Create a shared (default) nearest network in PCA space (or directly on matrix)
- Cluster on nearest network with Leiden or Louvain (k-means and hclust are alternatives)
seqfish_mini <- createNearestNetwork(gobject = seqfish_mini,
dimensions_to_use = 1:5,
k = 5)
seqfish_mini <- doLeidenCluster(gobject = seqfish_mini,
resolution = 0.4,
n_iterations = 1000)
# visualize UMAP cluster results
plotUMAP(gobject = seqfish_mini,
cell_color = "leiden_clus",
show_NN_network = TRUE,
point_size = 2.5)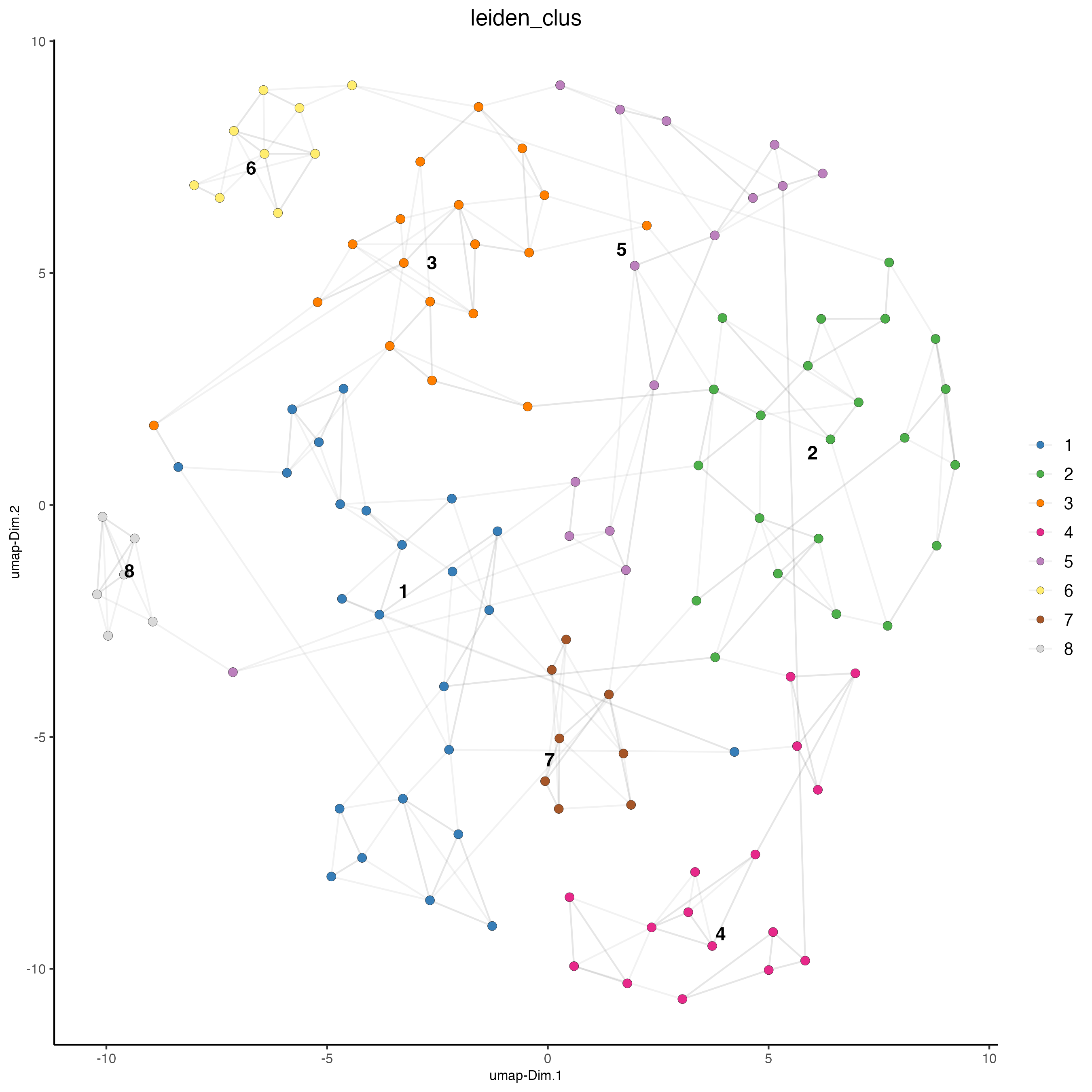
# visualize UMAP and spatial results
spatDimPlot(gobject = seqfish_mini,
cell_color = "leiden_clus",
spat_point_shape = "voronoi")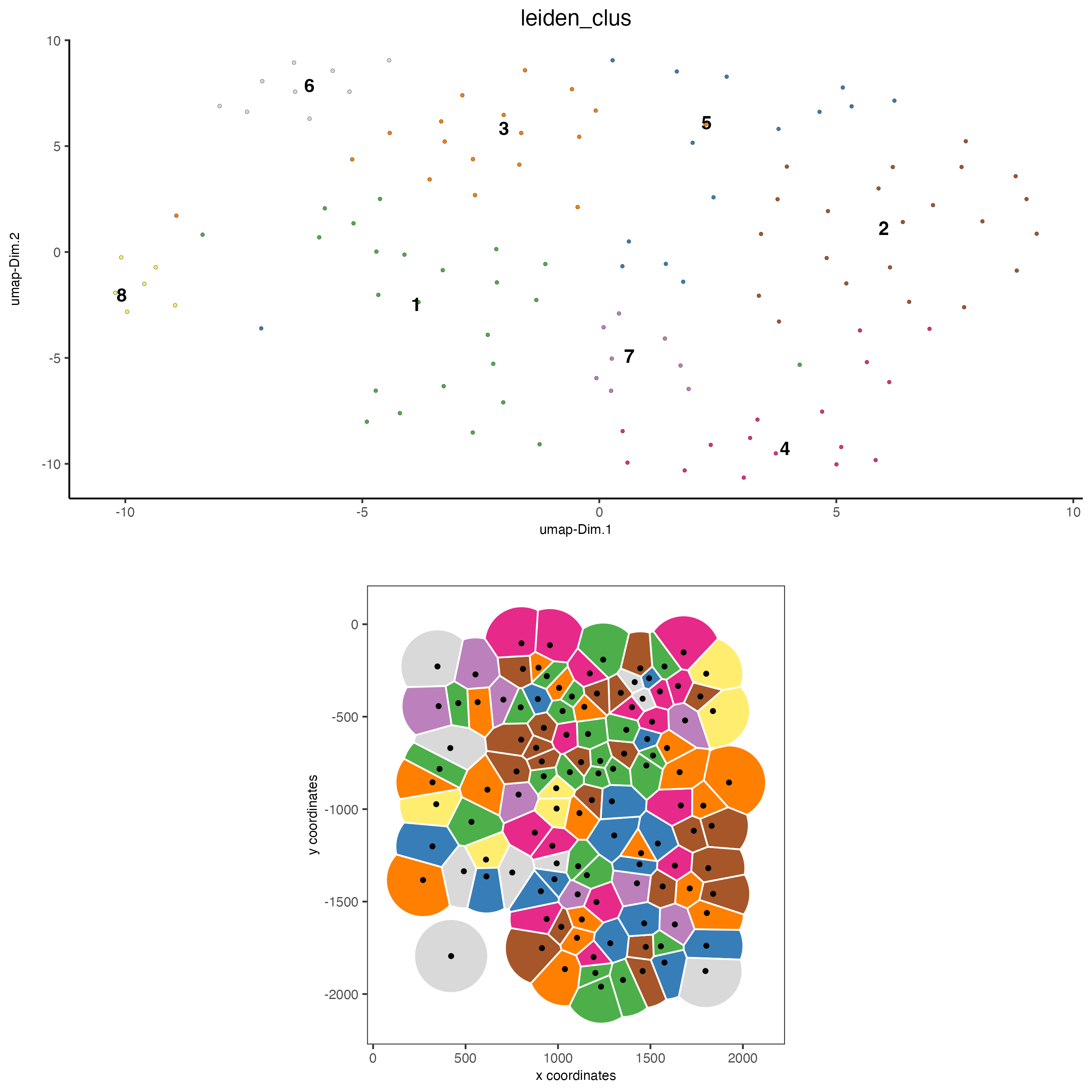
# heatmap and dendrogram
showClusterHeatmap(gobject = seqfish_mini,
cluster_column = "leiden_clus")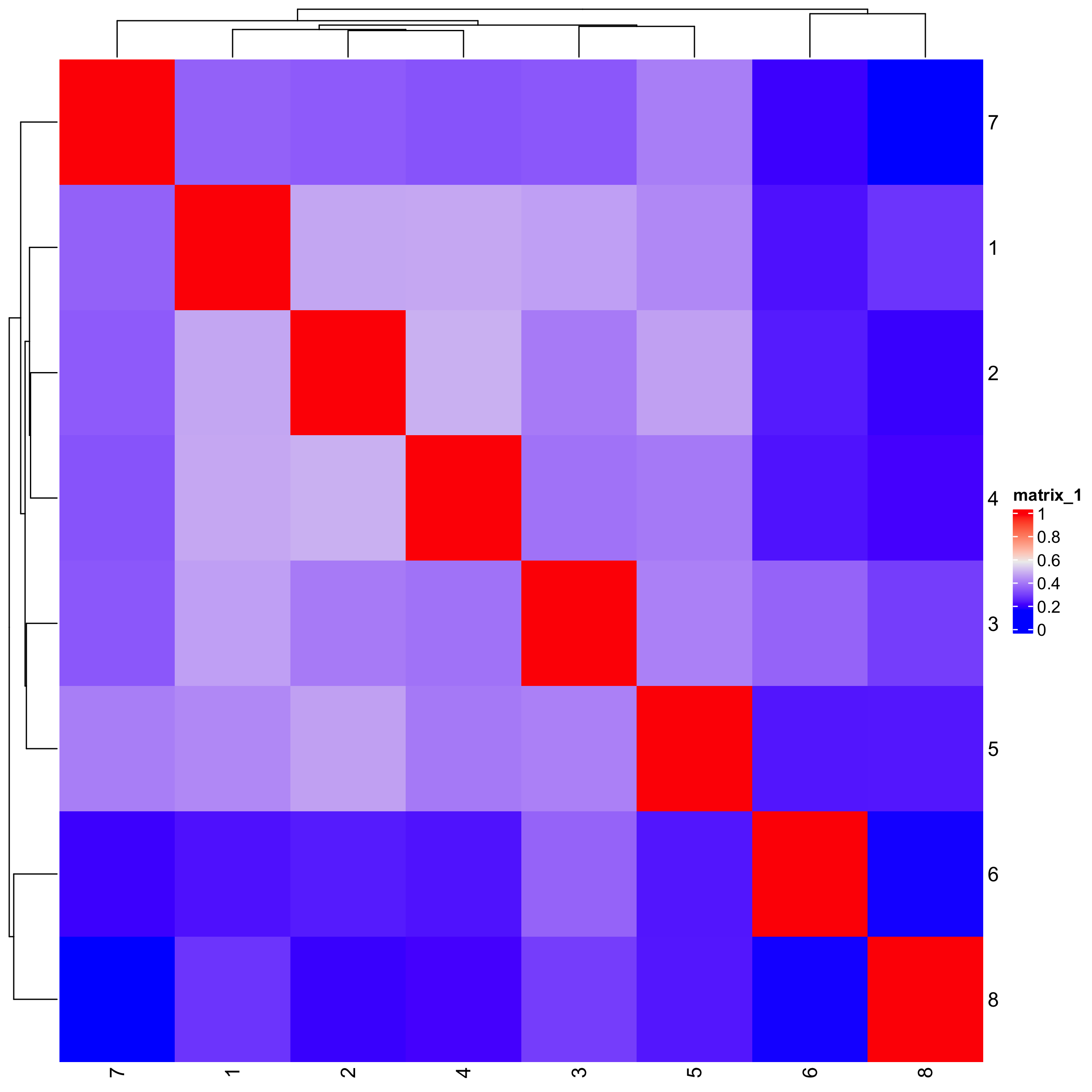
The following step requires the installation of {ggdendro}.
# install.packages("ggdendro")
library(ggdendro)
showClusterDendrogram(seqfish_mini,
h = 0.5,
rotate = TRUE,
cluster_column = "leiden_clus")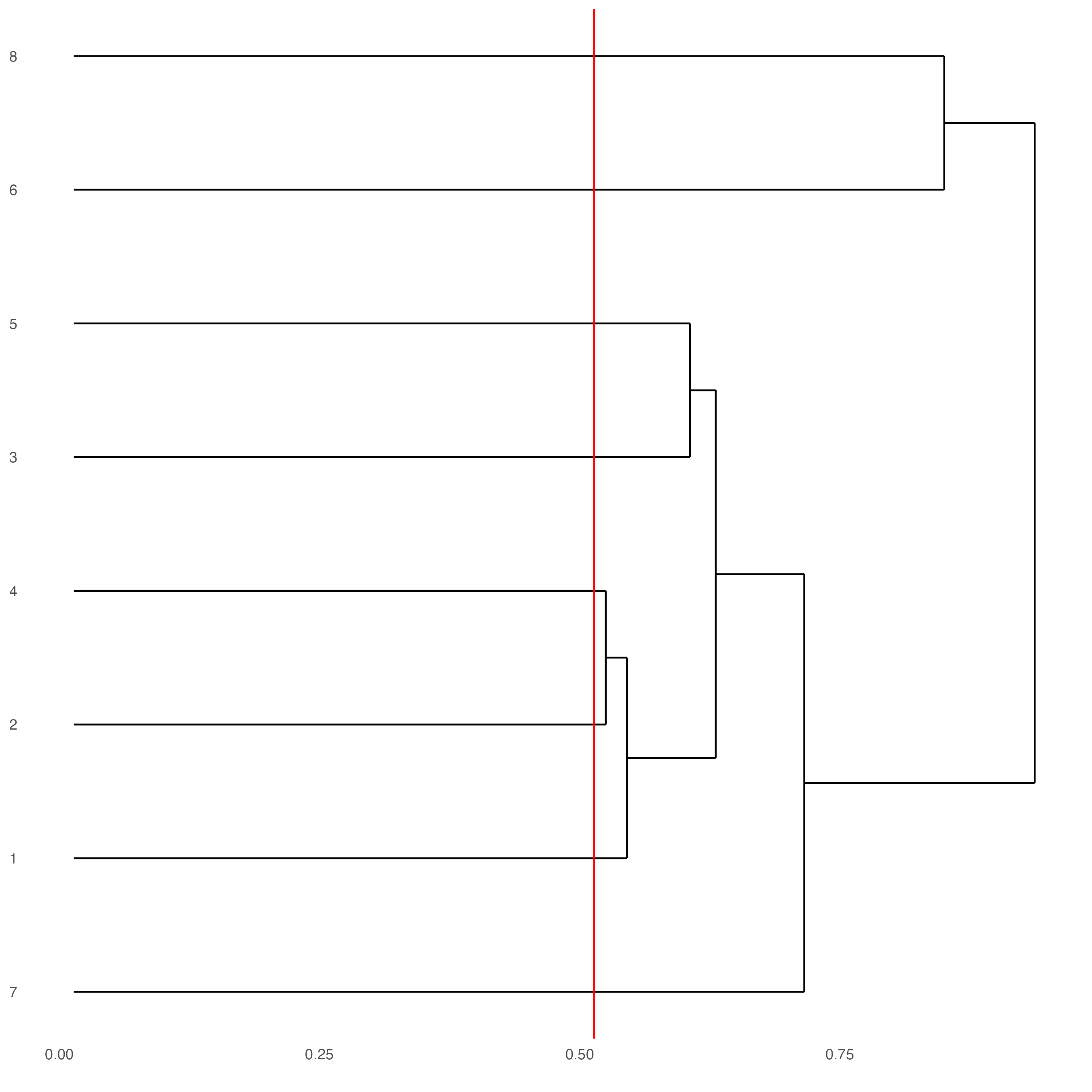
6 Differential expression
markers_gini <- findMarkers_one_vs_all(gobject = seqfish_mini,
method = "gini",
expression_values = "normalized",
cluster_column = "leiden_clus",
min_feats = 20,
min_expr_gini_score = 0.5,
min_det_gini_score = 0.5)
# get top 2 genes per cluster and visualize with violin plot
topgenes_gini = markers_gini[, head(.SD, 2), by = "cluster"]
violinPlot(seqfish_mini,
feats = topgenes_gini$feats[1:4],
cluster_column = "leiden_clus")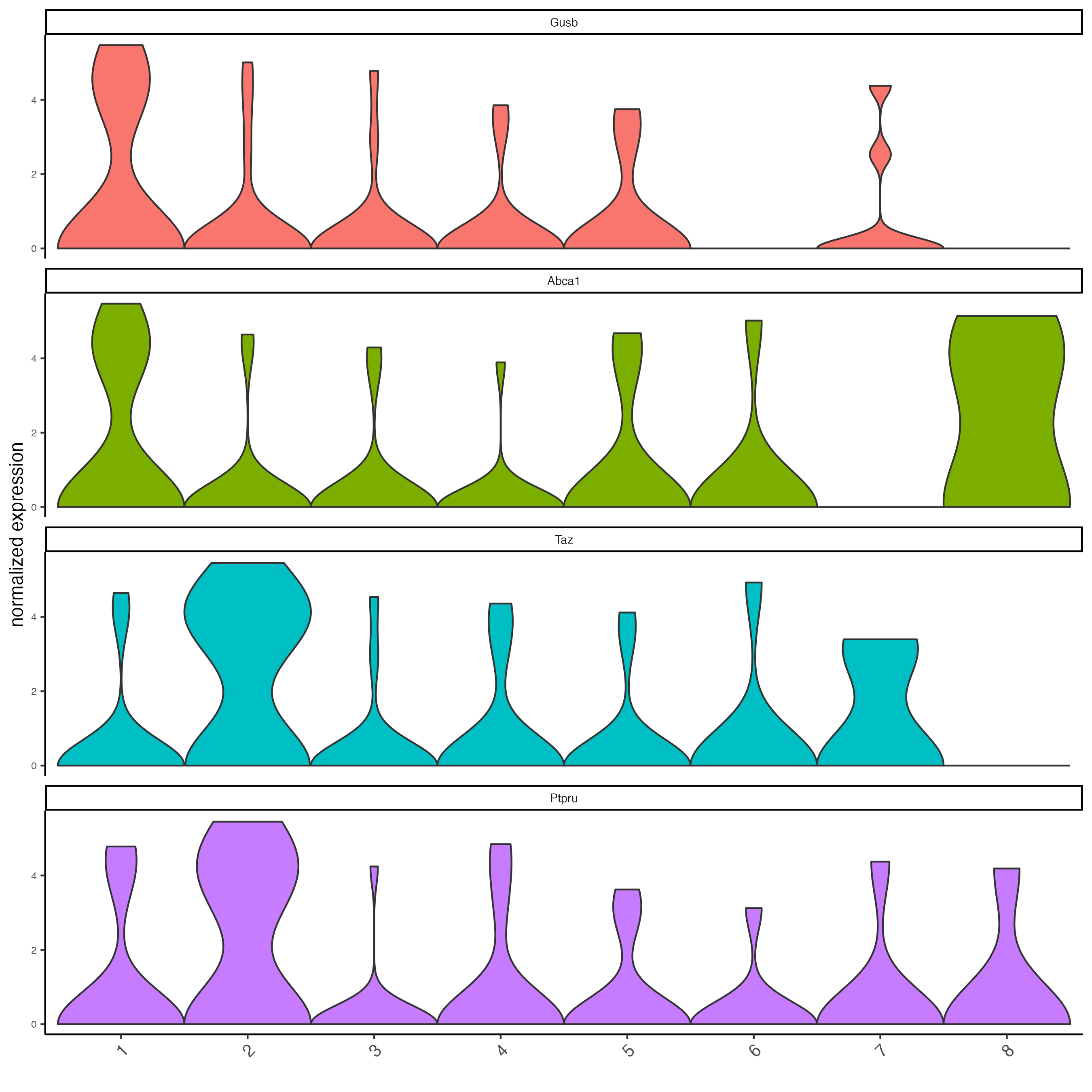
# get top 6 genes per cluster and visualize with heatmap
topgenes_gini <- markers_gini[, head(.SD, 6), by = "cluster"]
plotMetaDataHeatmap(seqfish_mini,
selected_feats = topgenes_gini$feats,
metadata_cols = "leiden_clus")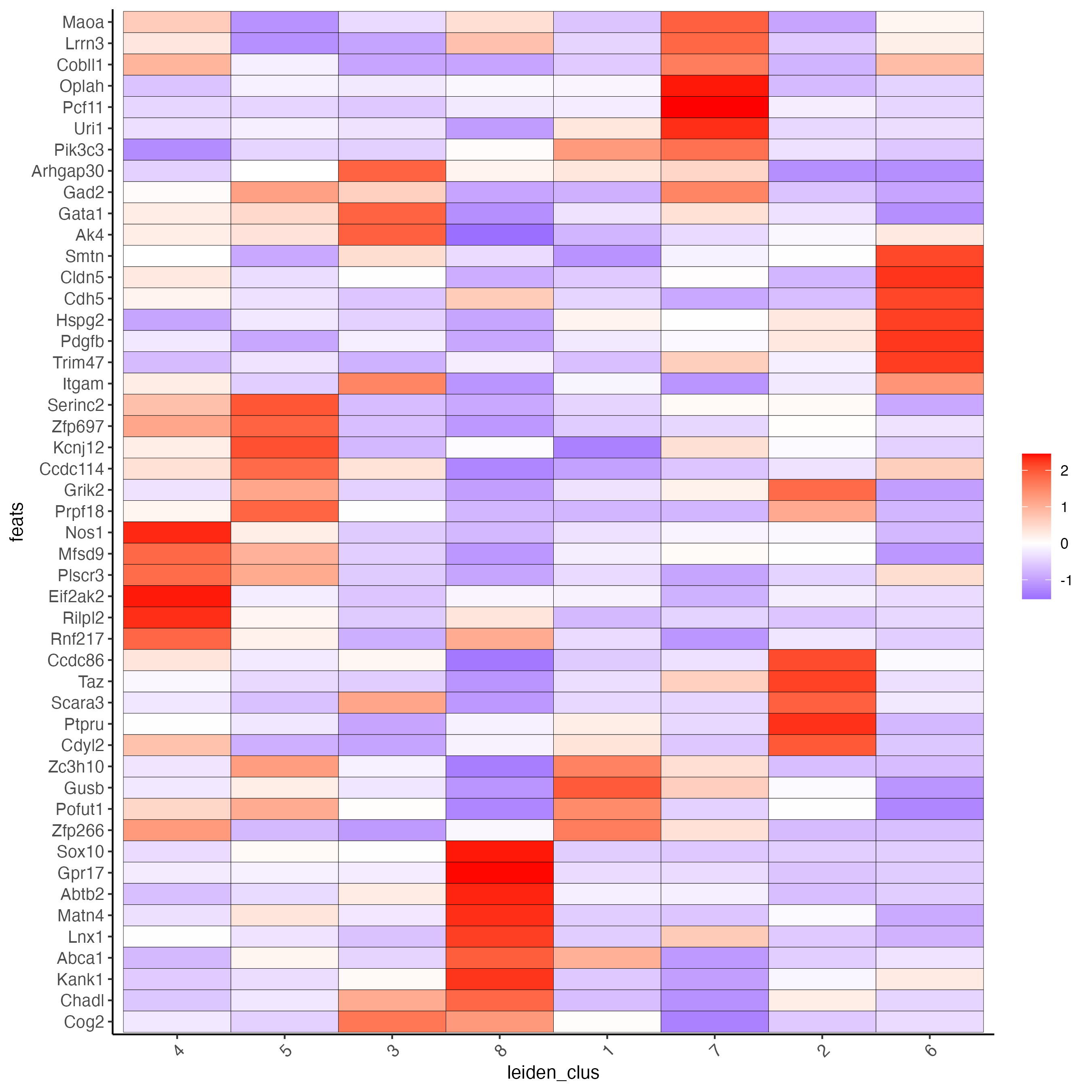
7 Cell type annotation
clusters_cell_types <- c("cell A", "cell B", "cell C", "cell D",
"cell E", "cell F", "cell G", "cell H")
names(clusters_cell_types) <- 1:8
seqfish_mini <- annotateGiotto(gobject = seqfish_mini,
annotation_vector = clusters_cell_types,
cluster_column = "leiden_clus",
name = "cell_types")
# check new cell metadata
pDataDT(seqfish_mini)
# visualize annotations
spatDimPlot(gobject = seqfish_mini,
cell_color = "cell_types",
spat_point_size = 3,
dim_point_size = 3)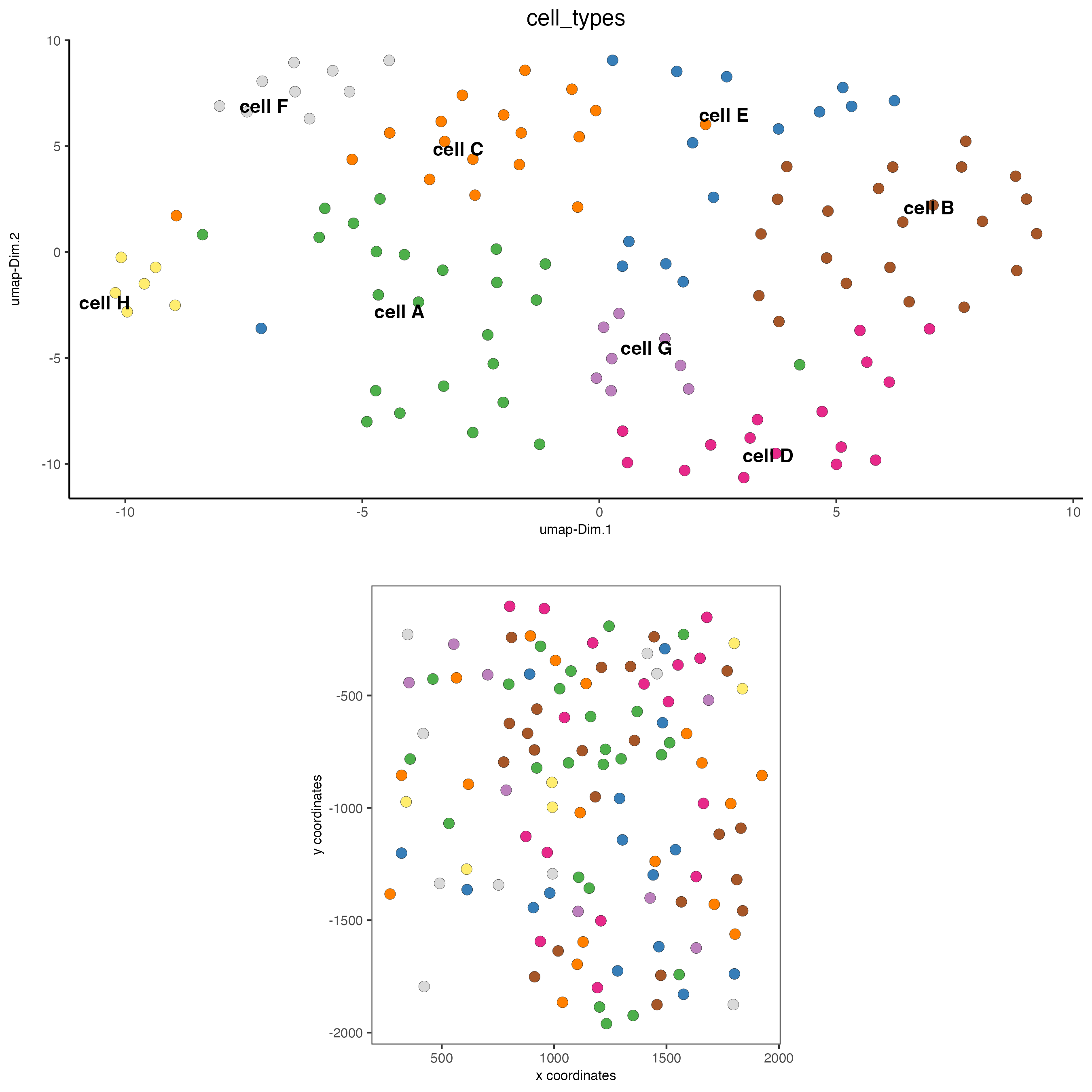
# heatmap
topgenes_heatmap <- markers_gini[, head(.SD, 4), by = "cluster"]
plotHeatmap(gobject = seqfish_mini,
feats = topgenes_heatmap$feats,
feat_order = "custom",
feat_custom_order = unique(topgenes_heatmap$feats),
cluster_column = "cell_types",
legend_nrows = 1)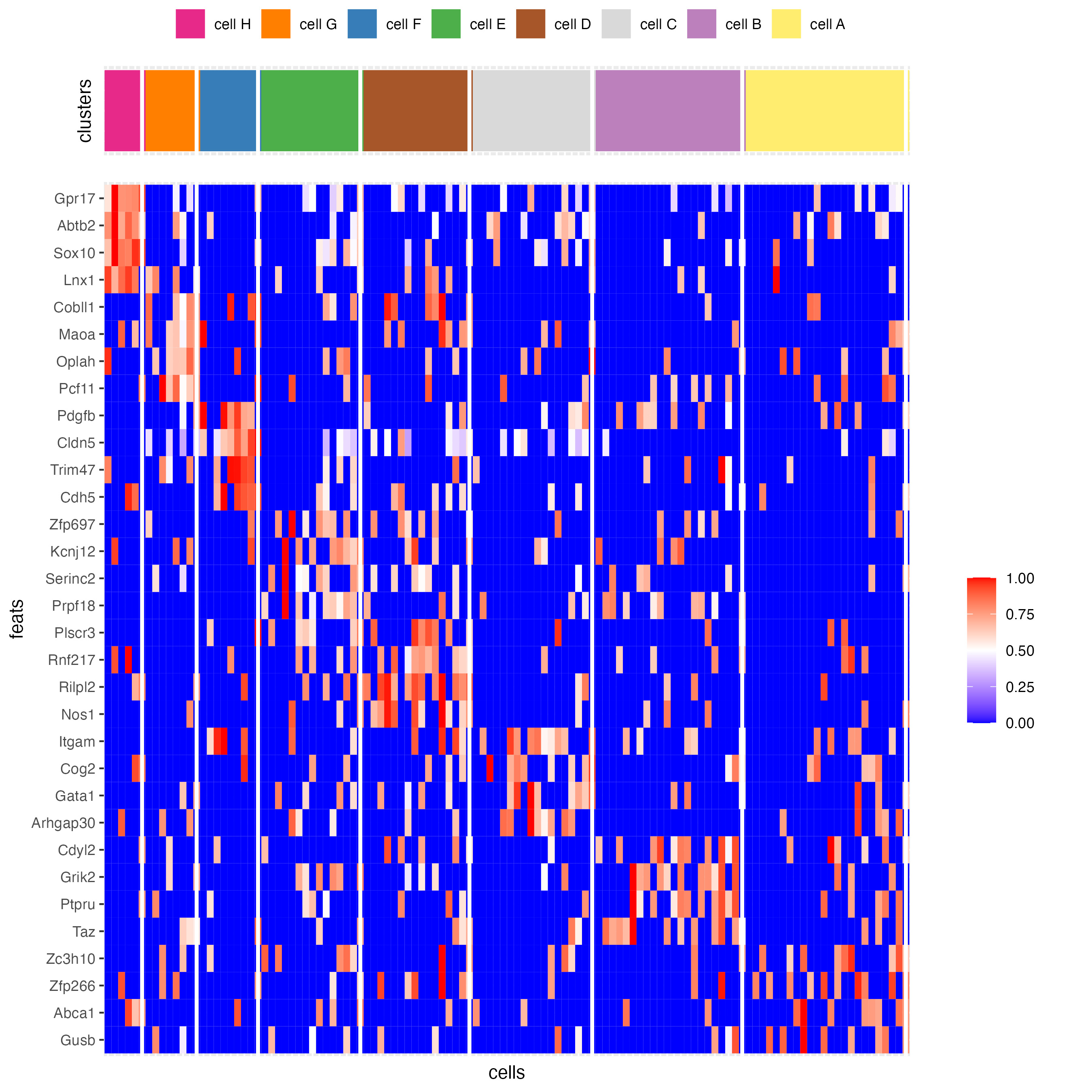
8 Spatial grid
- Create a grid based on defined step sizes in the x,y(,z) axes.
seqfish_mini <- createSpatialGrid(gobject = seqfish_mini,
sdimx_stepsize = 300,
sdimy_stepsize = 300,
minimum_padding = 50)
showGiottoSpatGrids(seqfish_mini)
# visualize grid
spatPlot(gobject = seqfish_mini,
show_grid = TRUE,
point_size = 1.5)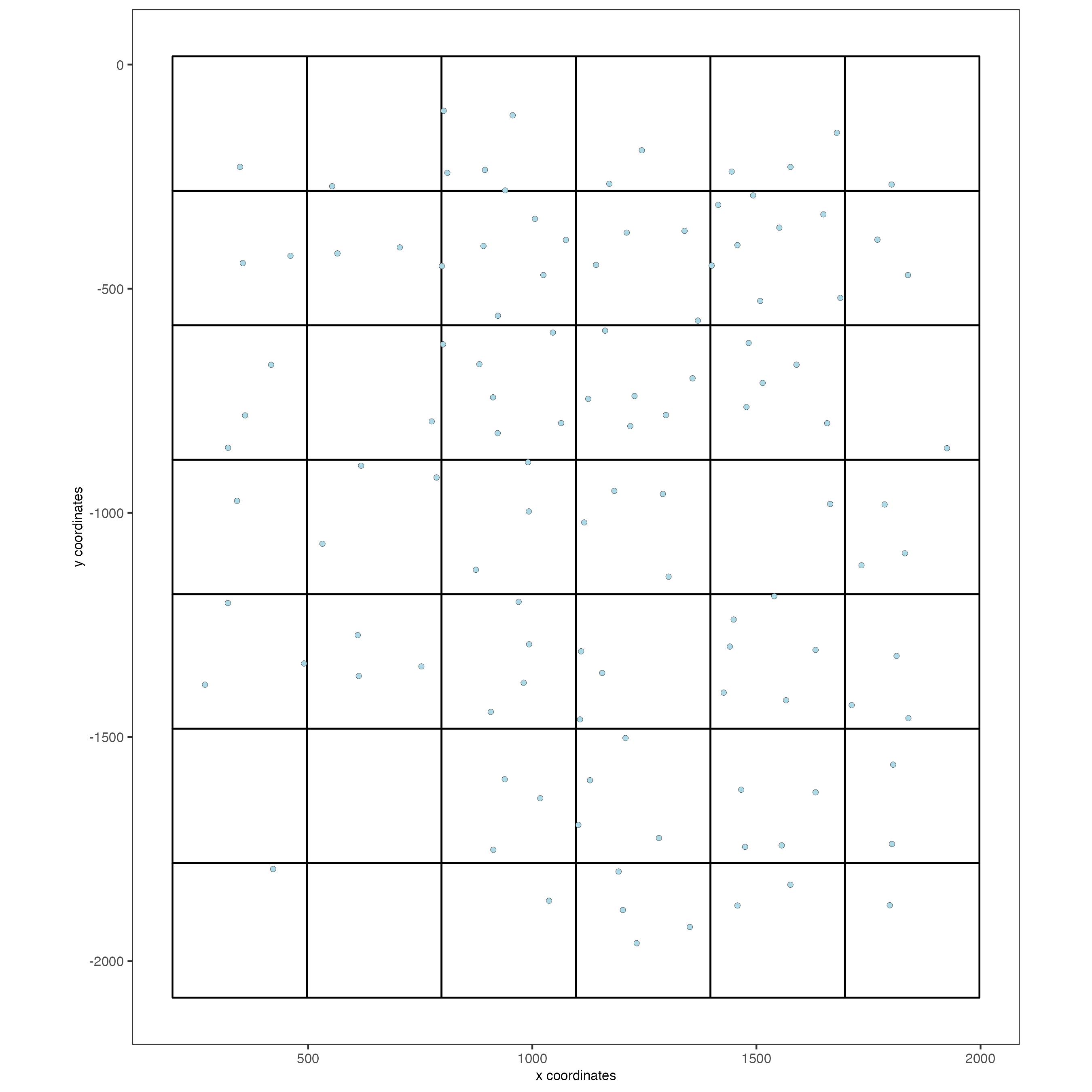
9 Spatial network
- Visualize information about the default Delaunay network
- Create a spatial Delaunay network (default)
- Create a spatial kNN network
plotStatDelaunayNetwork(gobject = seqfish_mini,
maximum_distance = 400)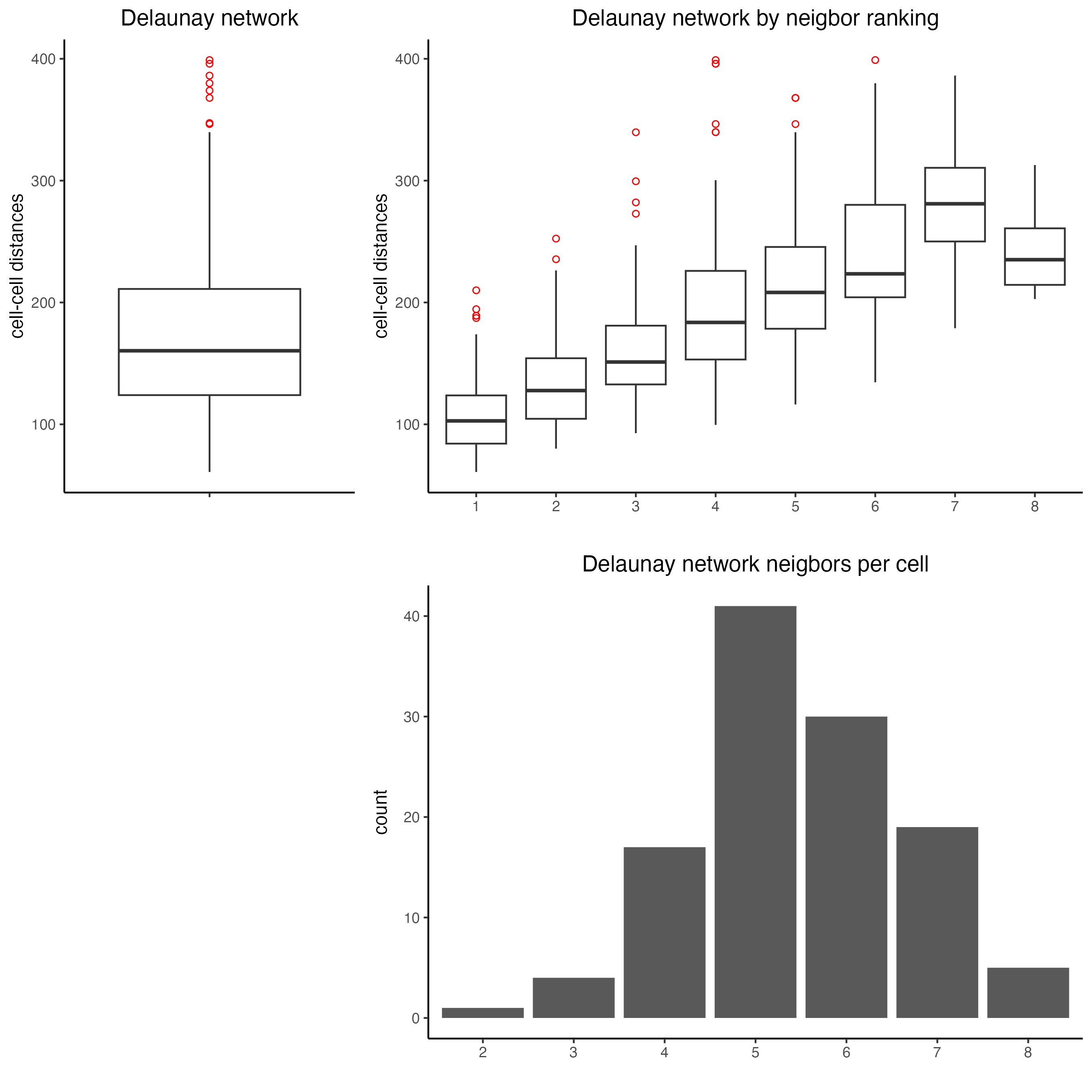
seqfish_mini <- createSpatialNetwork(gobject = seqfish_mini,
minimum_k = 2,
maximum_distance_delaunay = 400)
seqfish_mini <- createSpatialNetwork(gobject = seqfish_mini,
minimum_k = 2,
method = "kNN",
k = 10)
showGiottoSpatNetworks(seqfish_mini)
# visualize the two different spatial networks
spatPlot(gobject = seqfish_mini,
show_network = TRUE,
network_color = "blue",
spatial_network_name = "Delaunay_network",
point_size = 2.5,
cell_color = "leiden_clus")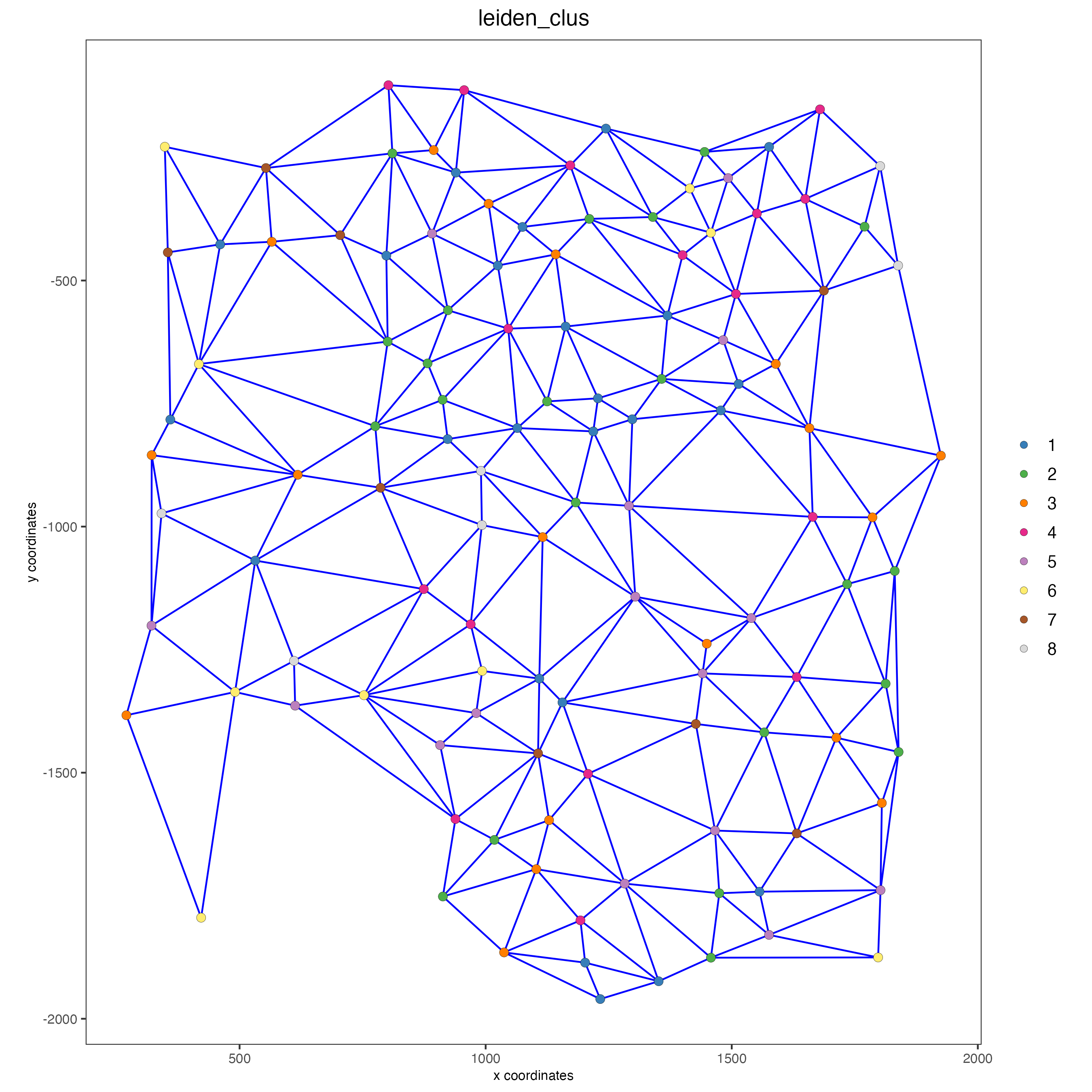
spatPlot(gobject = seqfish_mini,
show_network = TRUE,
network_color = "blue",
spatial_network_name = "kNN_network",
point_size = 2.5,
cell_color = "leiden_clus")
10 Spatial genes
Identify spatial genes with 3 different methods:
- binSpect with k-means binarization (default)
- binSpect with rank binarization
- silhouetteRank
Visualize top 4 genes per method.
km_spatialfeats <- binSpect(seqfish_mini)
spatFeatPlot2D(seqfish_mini,
expression_values = "scaled",
feats = km_spatialfeats[1:4]$feats,
point_shape = "border",
point_border_stroke = 0.1,
show_network = FALSE,
network_color = "lightgrey",
point_size = 2.5,
cow_n_col = 2)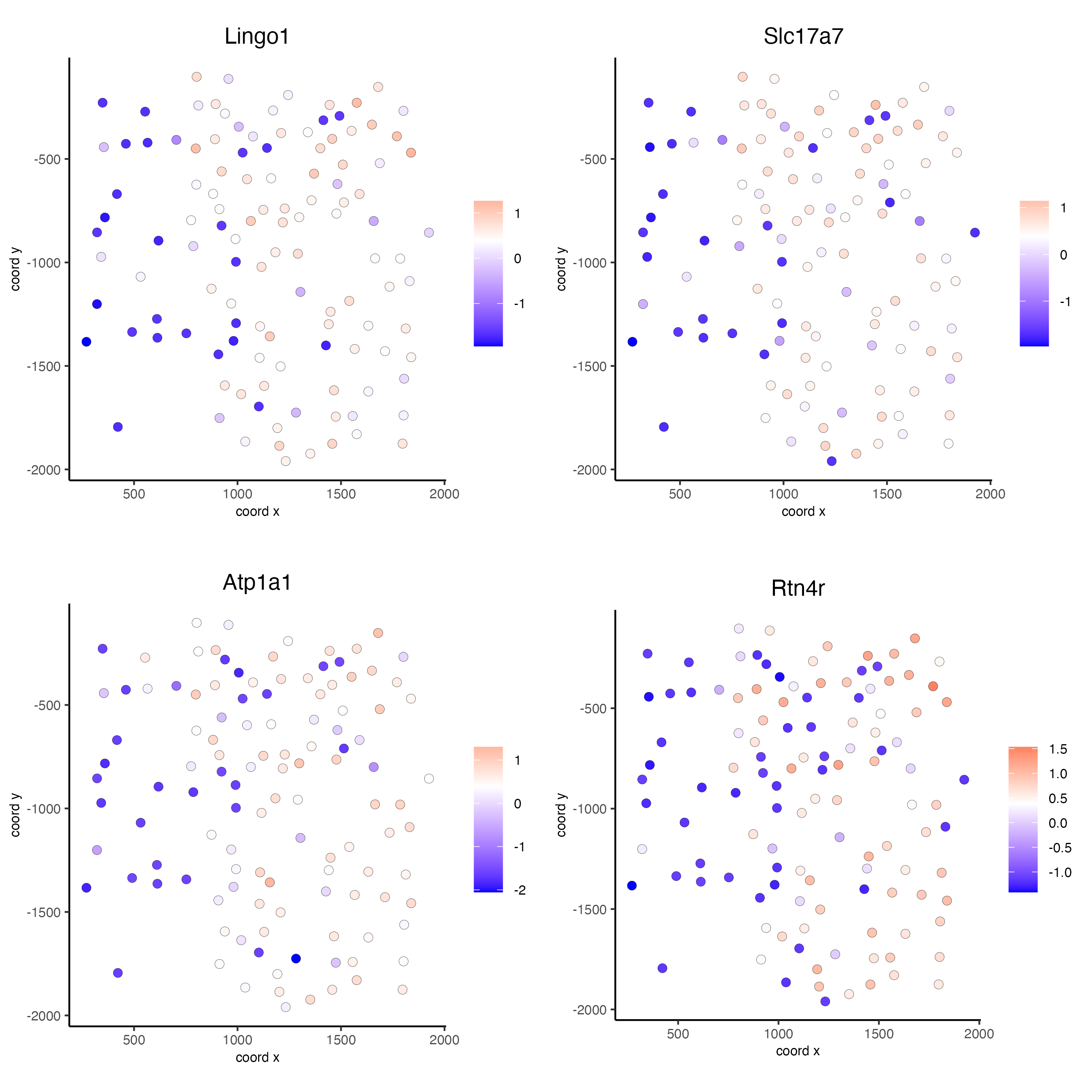
rank_spatialgenes <- binSpect(seqfish_mini,
bin_method = "rank")
spatFeatPlot2D(seqfish_mini,
expression_values = "scaled",
feats = rank_spatialgenes[1:4]$feats,
point_shape = "border",
point_border_stroke = 0.1,
show_network = FALSE,
network_color = "lightgrey",
point_size = 2.5,
cow_n_col = 2)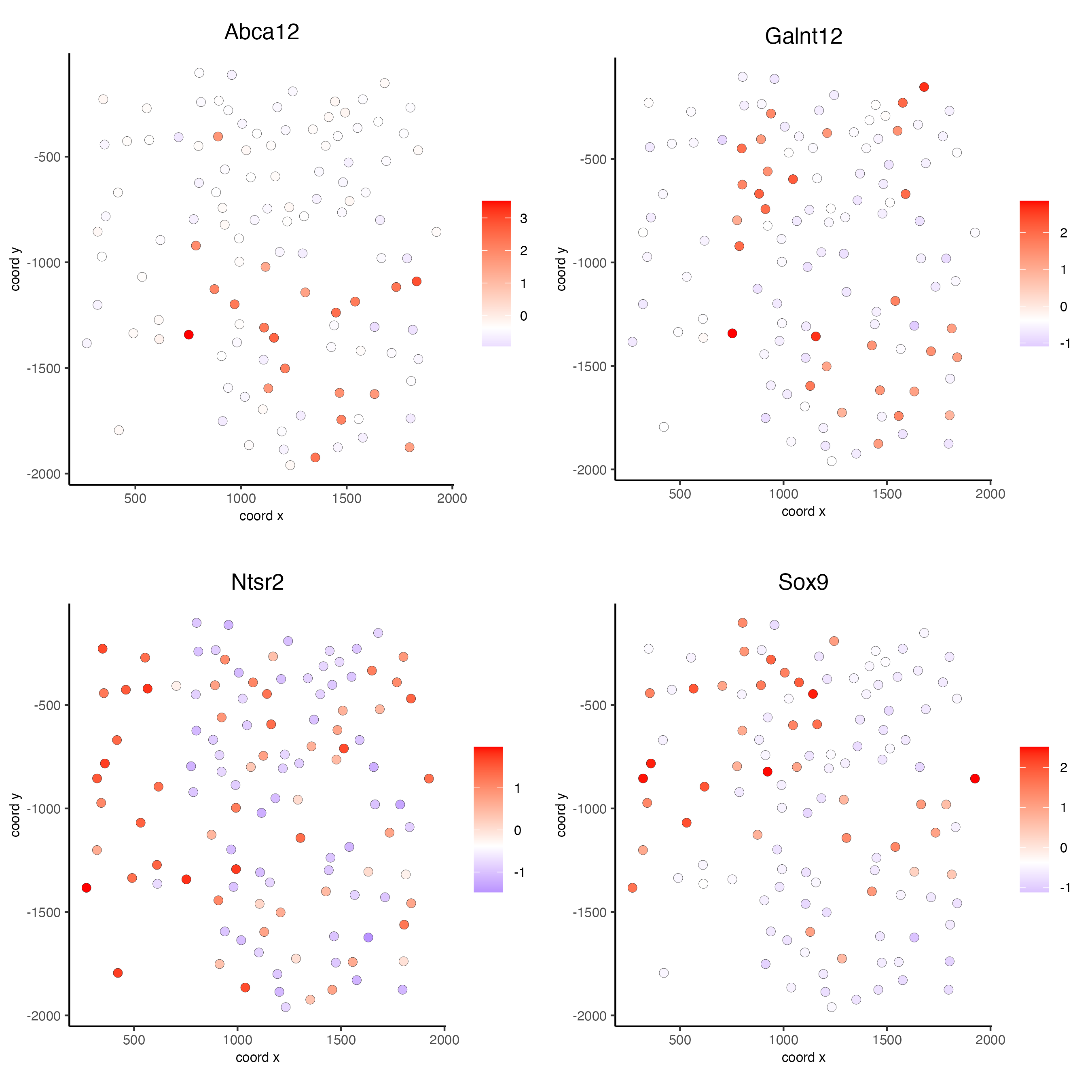
silh_spatialgenes <- silhouetteRank(gobject = seqfish_mini) # TODO: suppress print output
spatFeatPlot2D(seqfish_mini,
expression_values = "scaled",
feats = silh_spatialgenes[1:4]$genes,
point_shape = "border",
point_border_stroke = 0.1,
show_network = FALSE,
network_color = "lightgrey",
point_size = 2.5,
cow_n_col = 2)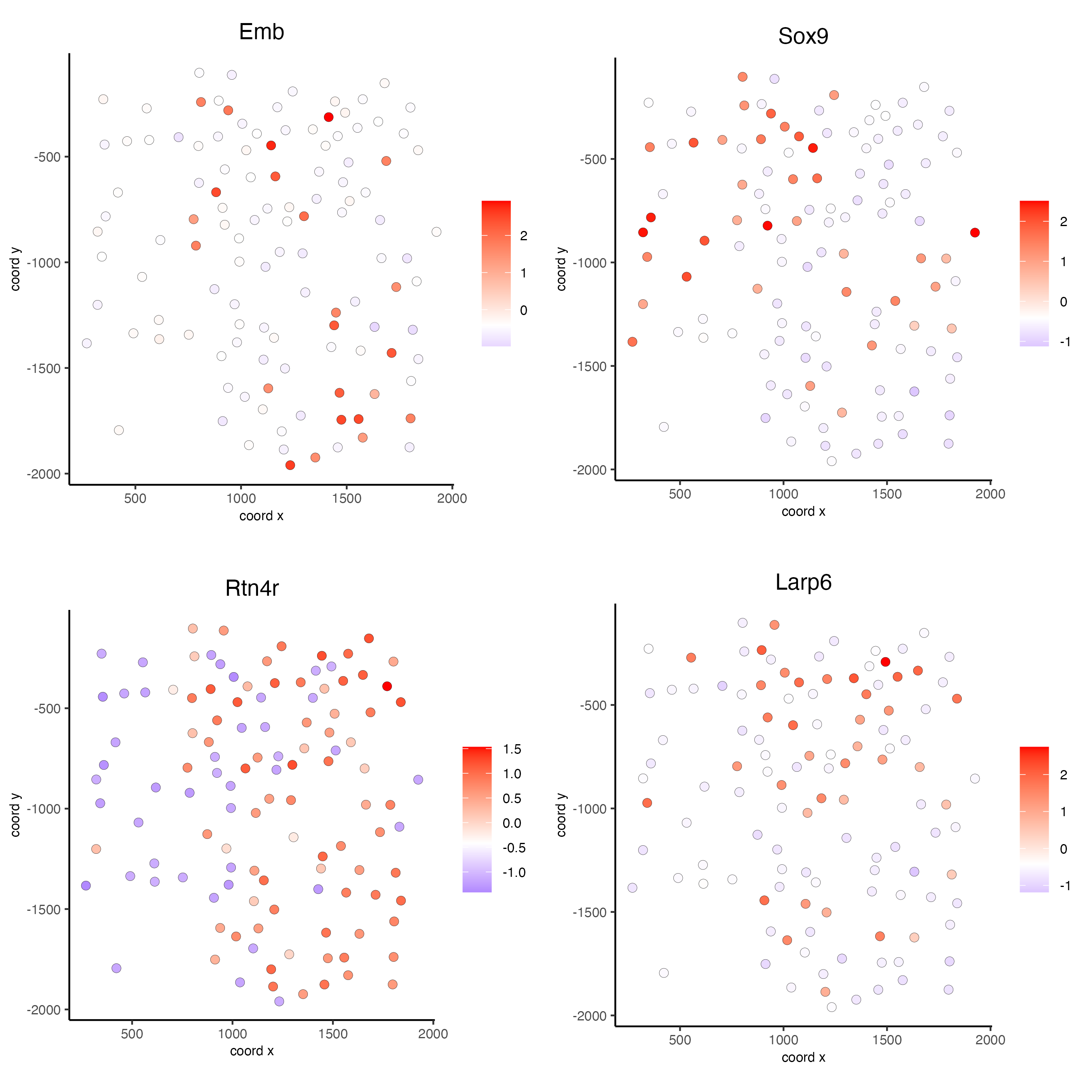
11 Spatial co-expression patterns
Identify robust spatial co-expression patterns using the spatial network or grid and a subset of individual spatial genes.
- Calculate spatial correlation scores
- Cluster correlation scores
# 1. calculate spatial correlation scores
ext_spatial_genes <- km_spatialfeats[1:500]$feats
spat_cor_netw_DT <- detectSpatialCorFeats(seqfish_mini,
method = "network",
spatial_network_name = "Delaunay_network",
subset_feats = ext_spatial_genes)
# 2. cluster correlation scores
spat_cor_netw_DT <- clusterSpatialCorFeats(spat_cor_netw_DT,
name = "spat_netw_clus",
k = 8)
heatmSpatialCorFeats(seqfish_mini,
spatCorObject = spat_cor_netw_DT,
use_clus_name = "spat_netw_clus")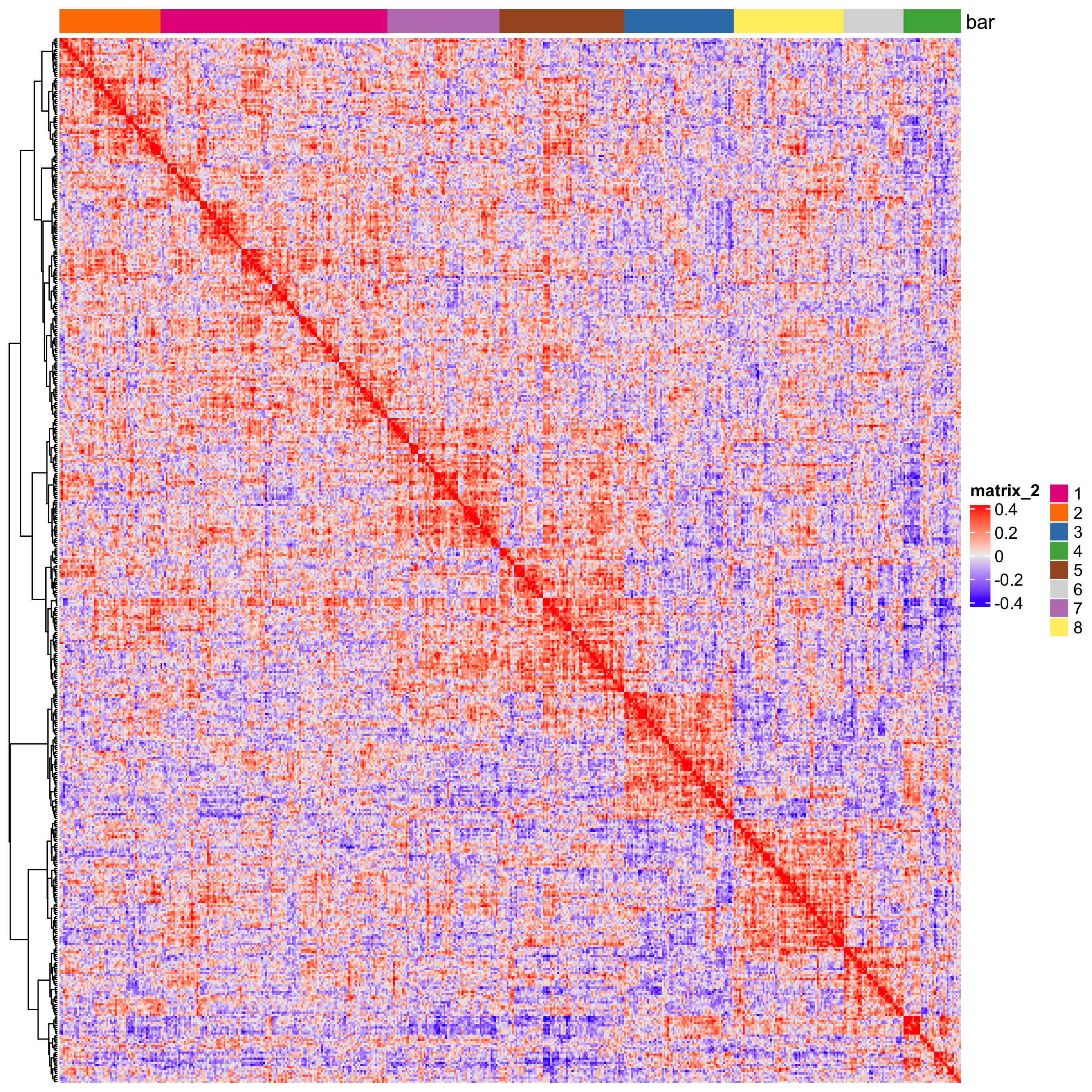
netw_ranks <- rankSpatialCorGroups(seqfish_mini,
spatCorObject = spat_cor_netw_DT,
use_clus_name = "spat_netw_clus")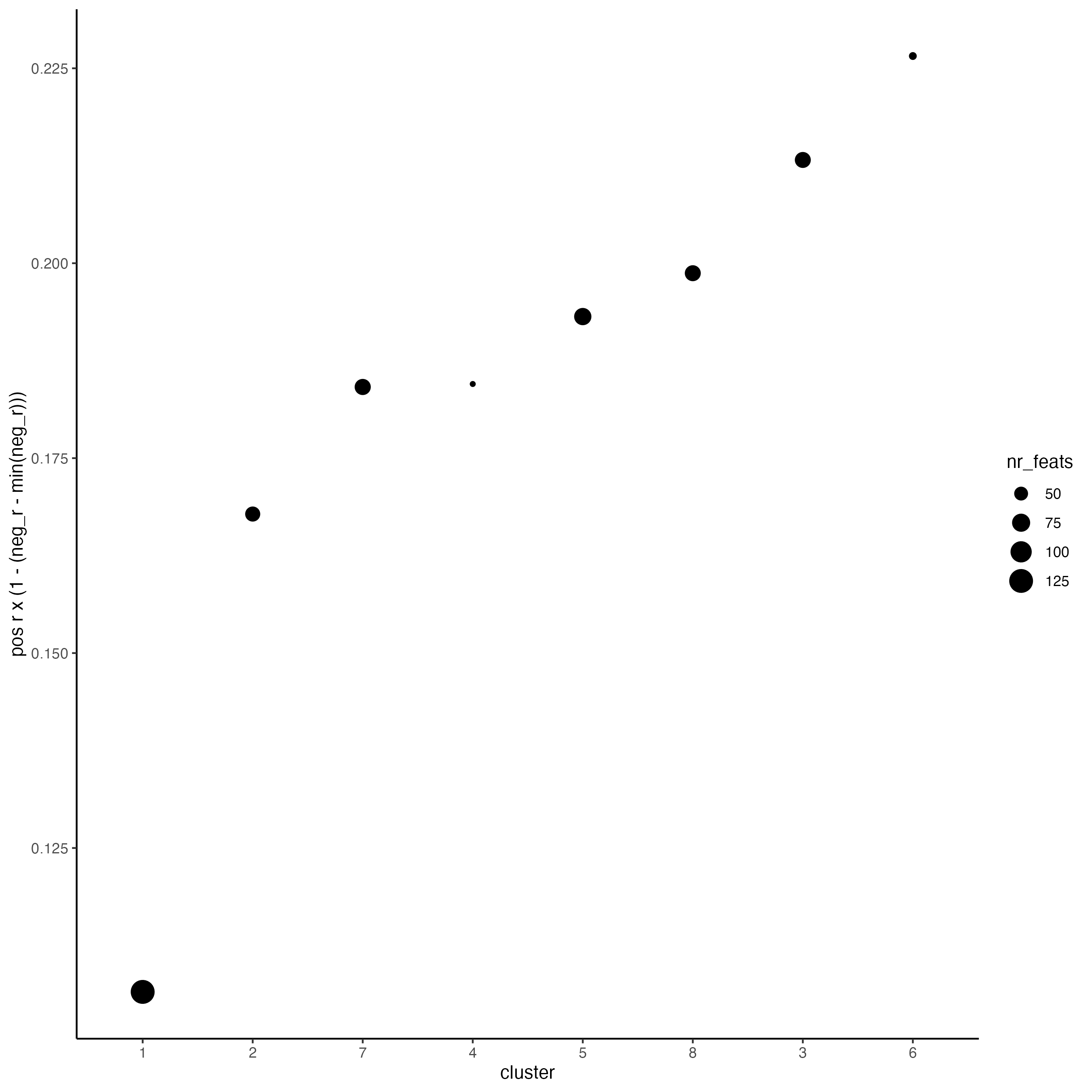
top_netw_spat_cluster <- showSpatialCorFeats(spat_cor_netw_DT,
use_clus_name = "spat_netw_clus",
selected_clusters = 6,
show_top_feats = 1)
cluster_genes_DT <- showSpatialCorFeats(spat_cor_netw_DT,
use_clus_name = "spat_netw_clus",
show_top_feats = 1)
cluster_genes <- cluster_genes_DT$clus
names(cluster_genes) <- cluster_genes_DT$feat_ID
seqfish_mini <- createMetafeats(seqfish_mini,
feat_clusters = cluster_genes,
name = "cluster_metagene")
spatCellPlot(seqfish_mini,
spat_enr_names = "cluster_metagene",
cell_annotation_values = netw_ranks$clusters,
point_size = 1.5,
cow_n_col = 3)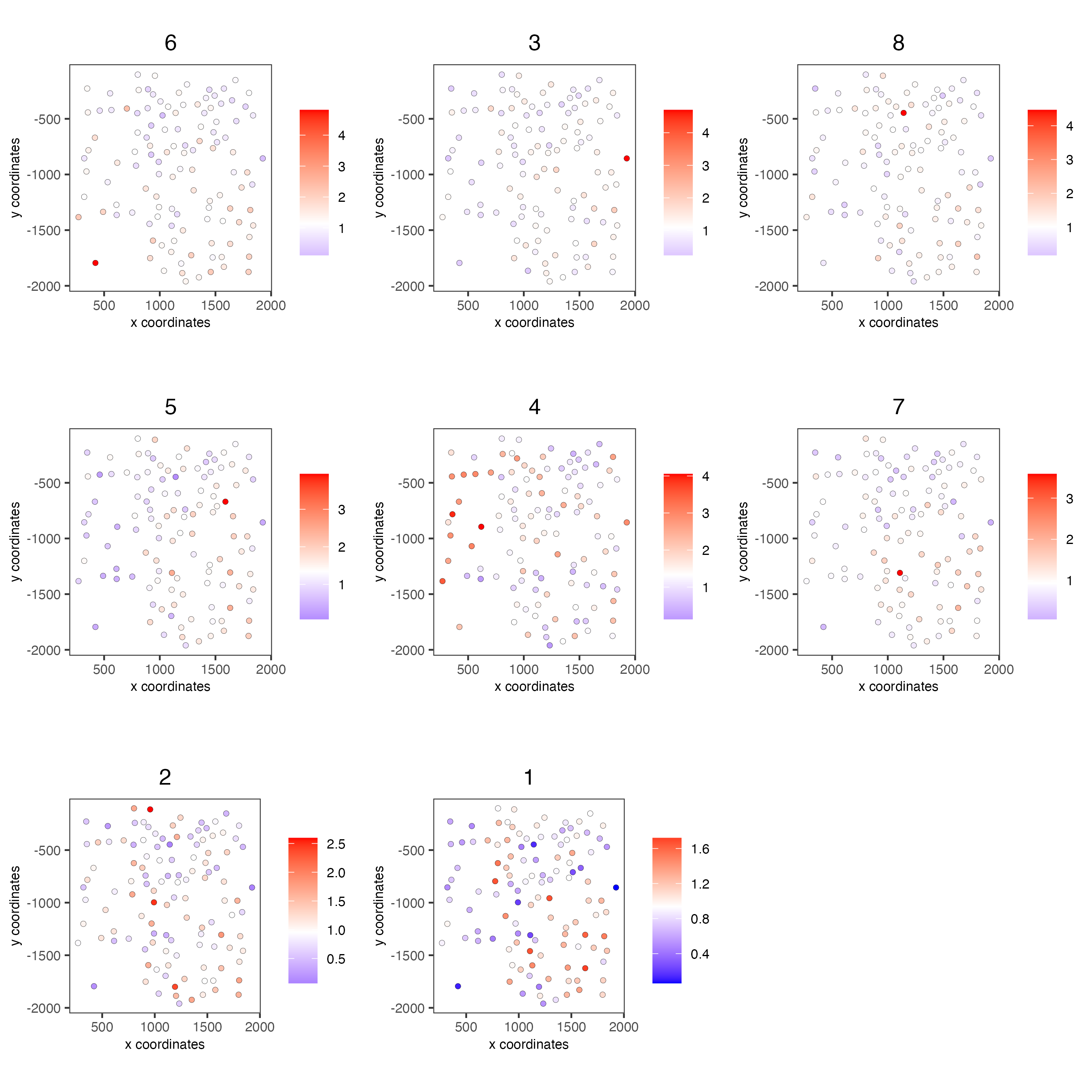
12 Spatial HMRF domains
The following HMRF function requires {smfishHmrf} .
# remotes::install_bitbucket(repo = "qzhudfci/smfishhmrf-r", ref="master")
library(smfishHmrf)
hmrf_folder <- paste0(results_folder, "/11_HMRF/")
if(!file.exists(hmrf_folder)) dir.create(hmrf_folder, recursive = TRUE)
# perform hmrf
my_spatial_genes <- km_spatialfeats[1:100]$feats
HMRF_spatial_genes <- doHMRF(gobject = seqfish_mini,
expression_values = "scaled",
spatial_genes = my_spatial_genes,
spatial_network_name = "Delaunay_network",
k = 9,
betas = c(28,2,2),
output_folder = paste0(hmrf_folder, "/", "Spatial_genes/SG_top100_k9_scaled"))
# check and select hmrf
for(i in seq(28, 30, by = 2)) {
viewHMRFresults2D(gobject = seqfish_mini,
HMRFoutput = HMRF_spatial_genes,
k = 9,
betas_to_view = i,
point_size = 2)
}
seqfish_mini <- addHMRF(gobject = seqfish_mini,
HMRFoutput = HMRF_spatial_genes,
k = 9,
betas_to_add = 28,
hmrf_name = "HMRF")
# visualize selected hmrf result
giotto_colors <- getDistinctColors(9)
names(giotto_colors) <- 1:9
spatPlot(gobject = seqfish_mini,
cell_color = "HMRF_k9_b.28",
point_size = 3,
coord_fix_ratio = 1,
cell_color_code = giotto_colors)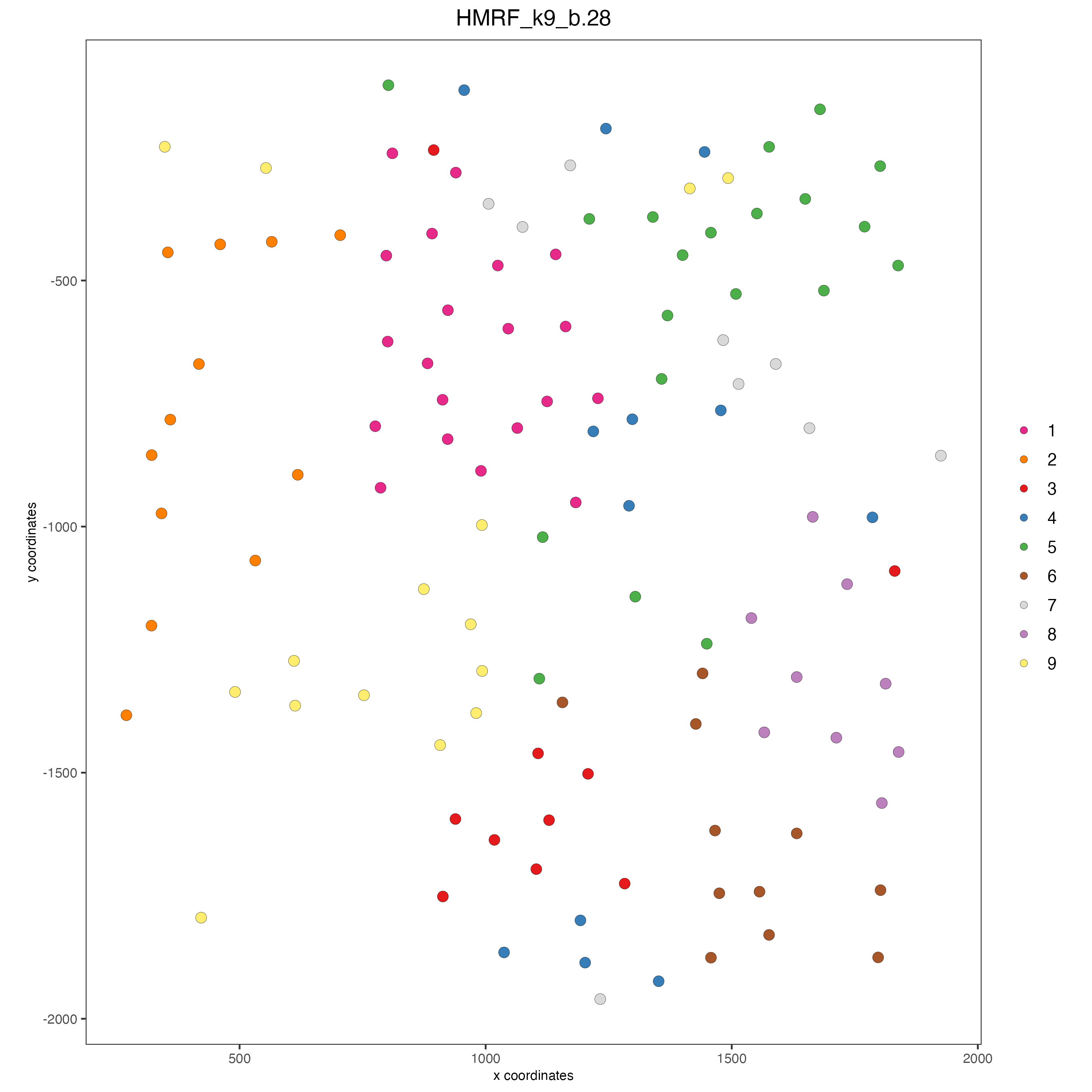
13 Cell neighborhood: cell-type/cell-type interactions
set.seed(seed = 2841)
cell_proximities <- cellProximityEnrichment(gobject = seqfish_mini,
cluster_column = "cell_types",
spatial_network_name = "Delaunay_network",
adjust_method = "fdr",
number_of_simulations = 1000)
# barplot
cellProximityBarplot(gobject = seqfish_mini,
CPscore = cell_proximities,
min_orig_ints = 5,
min_sim_ints = 5,
p_val = 0.5)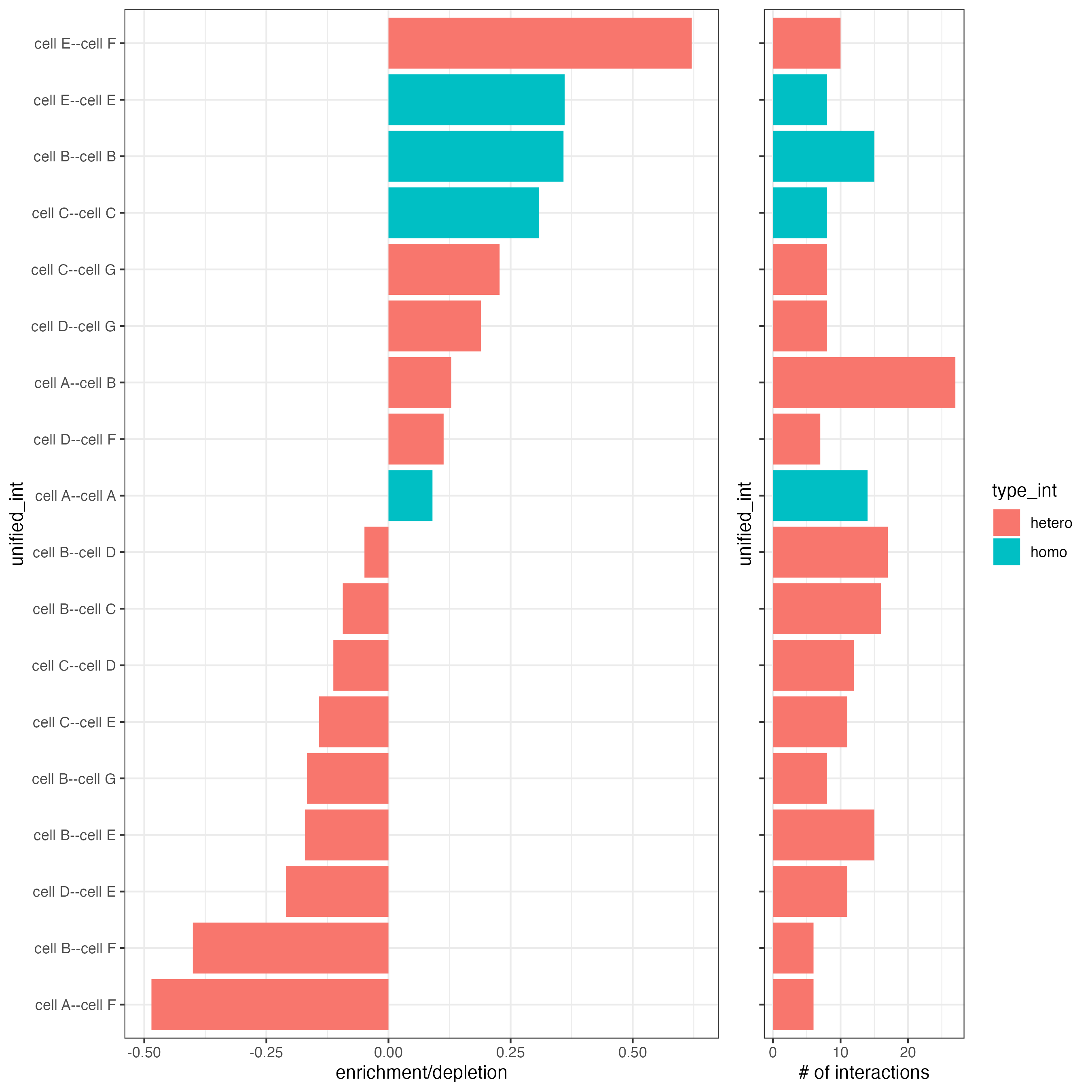
## heatmap
cellProximityHeatmap(gobject = seqfish_mini,
CPscore = cell_proximities,
order_cell_types = TRUE,
scale = TRUE,
color_breaks = c(-1.5, 0, 1.5),
color_names = c("blue", "white", "red"))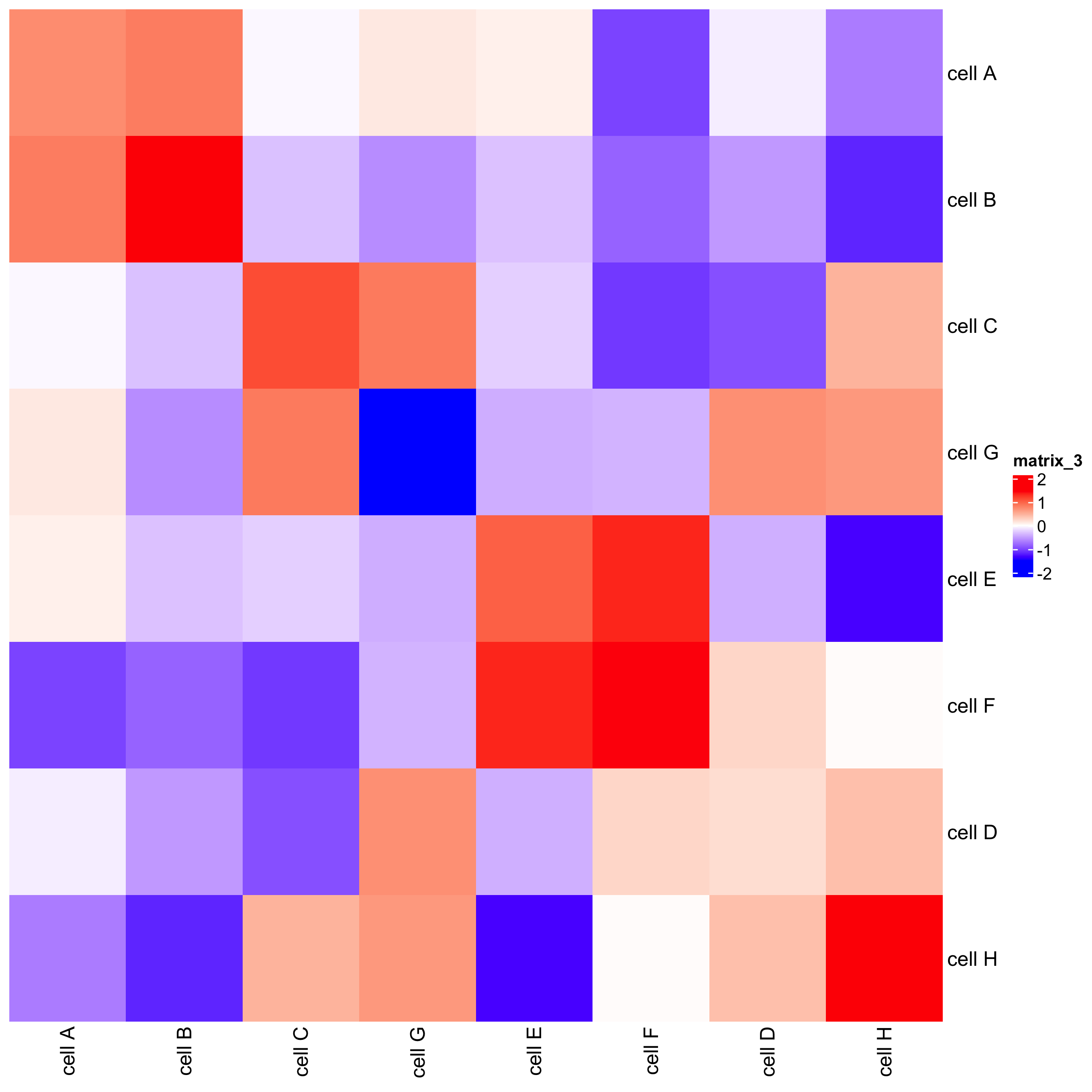
# network
cellProximityNetwork(gobject = seqfish_mini,
CPscore = cell_proximities,
remove_self_edges = TRUE,
only_show_enrichment_edges = TRUE)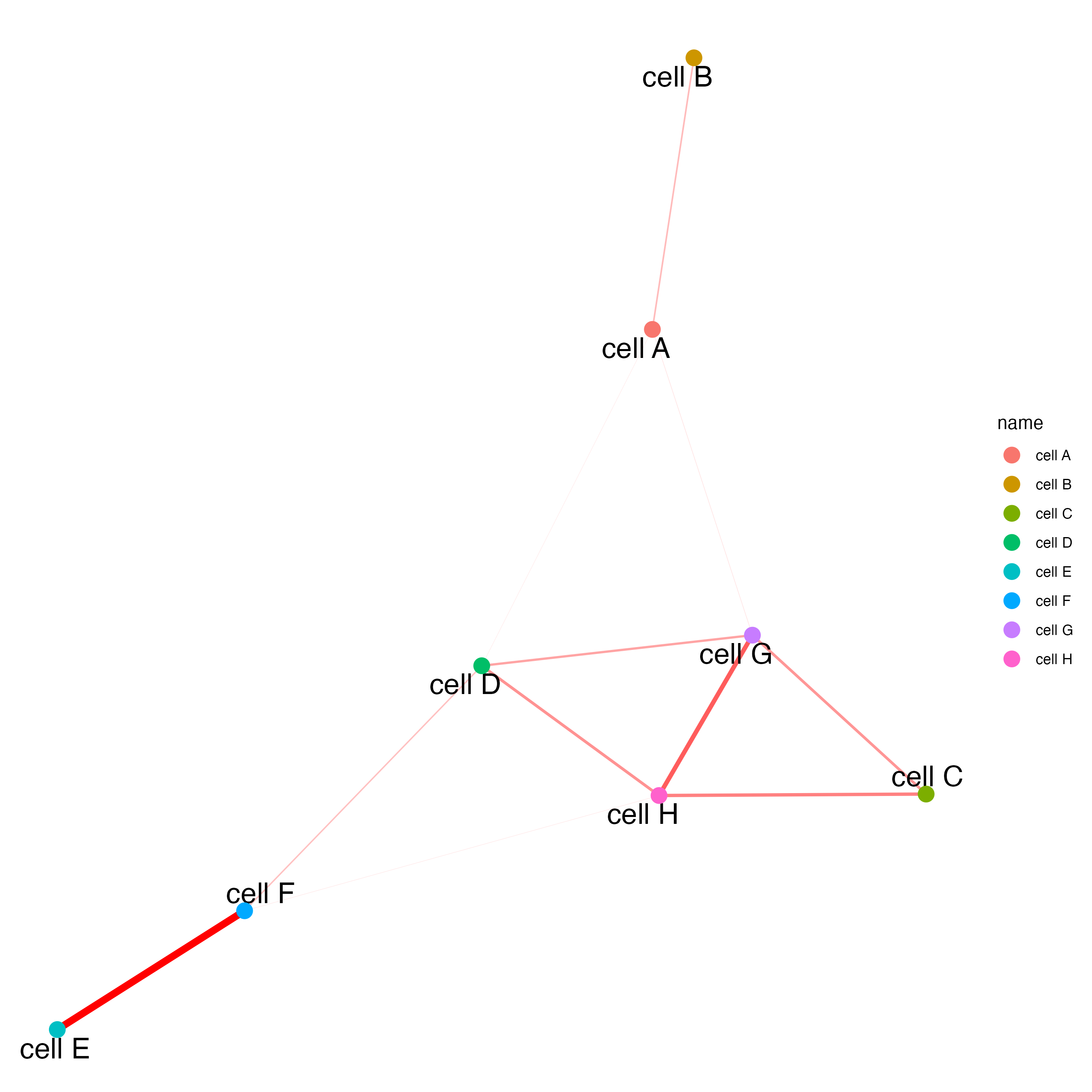
# network with self-edges
cellProximityNetwork(gobject = seqfish_mini,
CPscore = cell_proximities,
remove_self_edges = FALSE,
self_loop_strength = 0.3,
only_show_enrichment_edges = FALSE,
rescale_edge_weights = TRUE,
node_size = 8,
edge_weight_range_depletion = c(1, 2),
edge_weight_range_enrichment = c(2,5))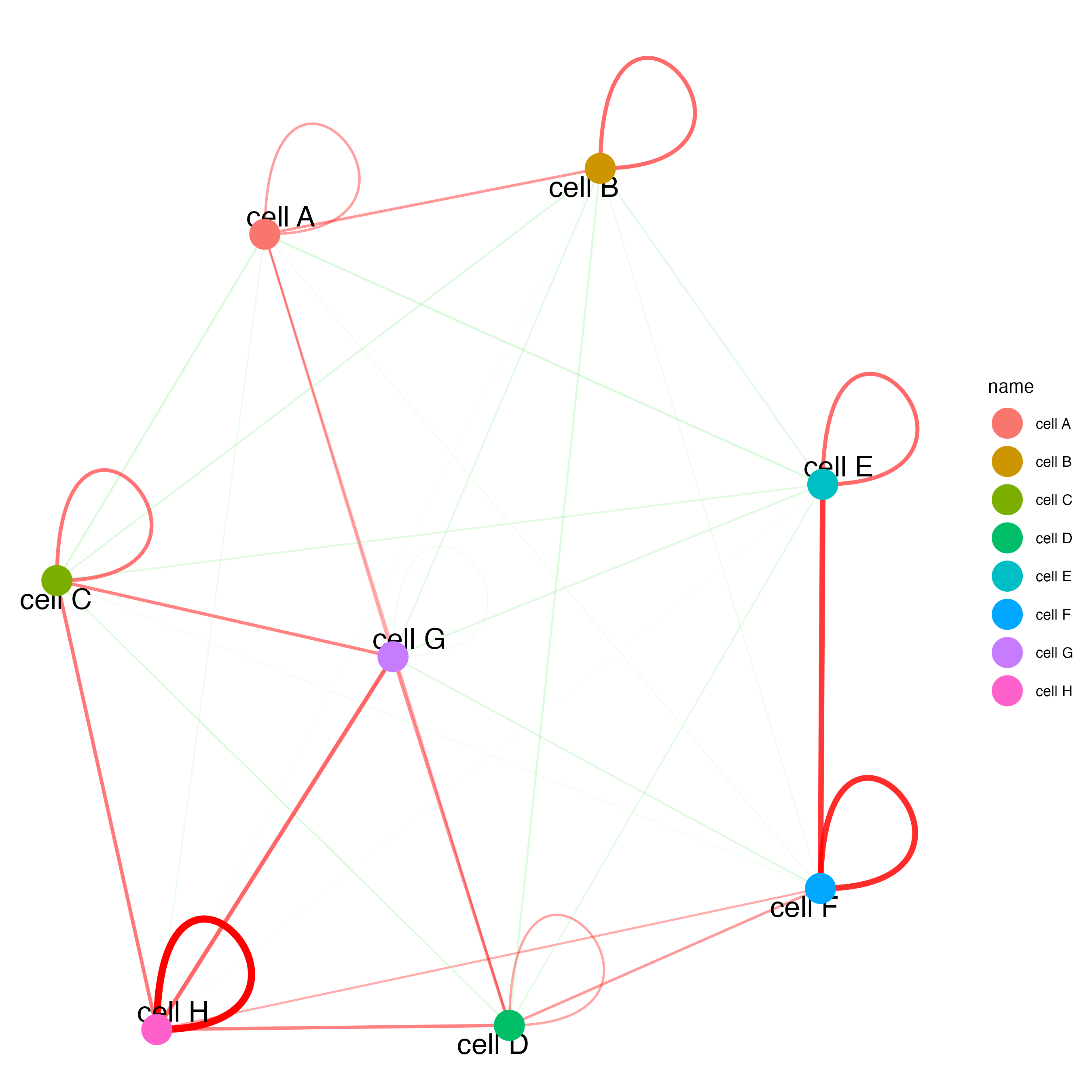
13.1 Visualization of specific cell types
# Option 1
spec_interaction <- "cell D--cell F"
cellProximitySpatPlot2D(gobject = seqfish_mini,
interaction_name = spec_interaction,
show_network = TRUE,
cluster_column = "cell_types",
cell_color = "cell_types",
cell_color_code = c("cell D" = "lightblue", "cell F" = "red"),
point_size_select = 4,
point_size_other = 2)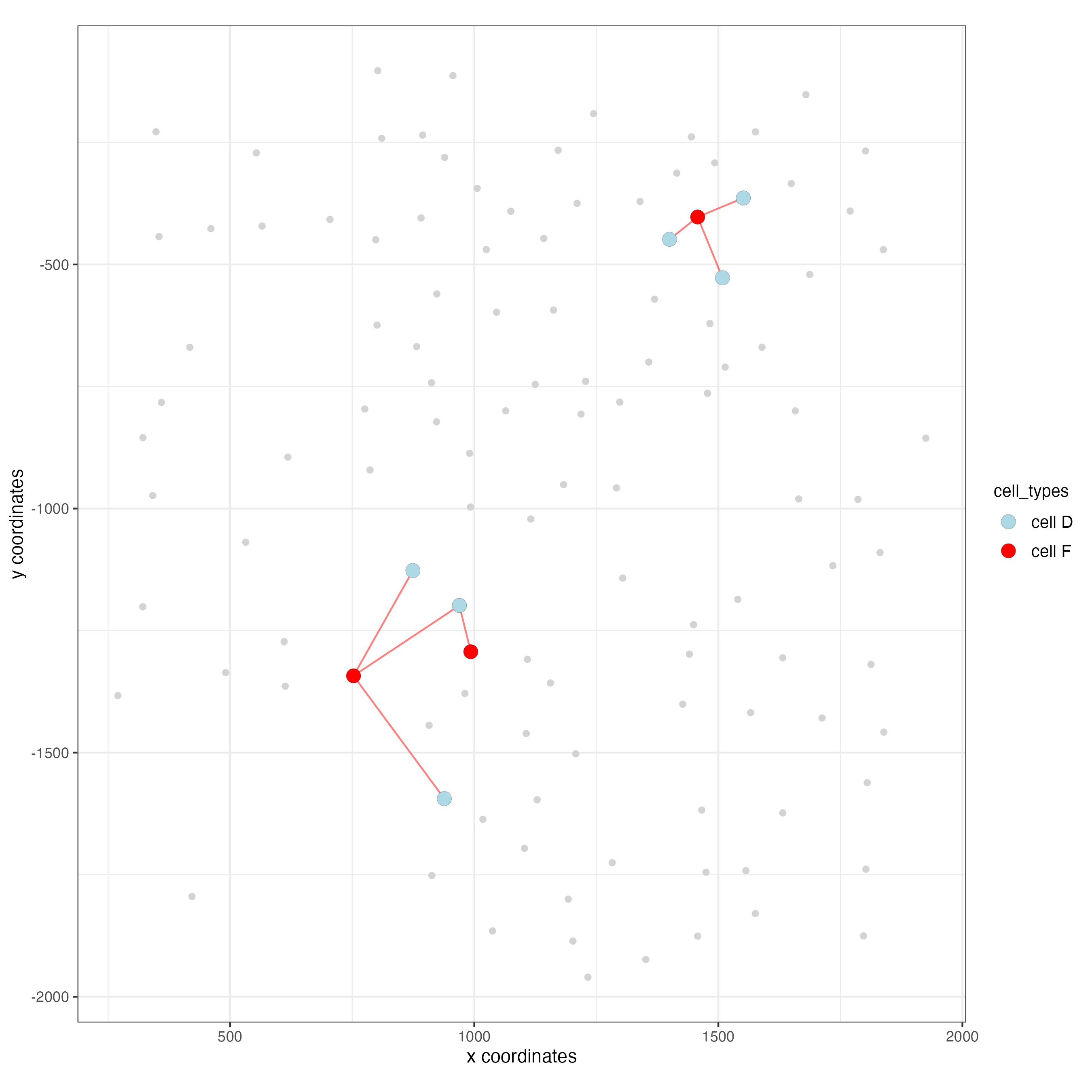
# Option 2: create additional metadata
seqfish_mini <- addCellIntMetadata(seqfish_mini,
spat_unit = "cell",
spatial_network = "Delaunay_network",
cluster_column = "cell_types",
cell_interaction = spec_interaction,
name = "D_F_interactions")
spatPlot(seqfish_mini,
cell_color = "D_F_interactions",
legend_symbol_size = 3,
select_cell_groups = c("other_cell D", "other_cell F", "select_cell D", "select_cell F"))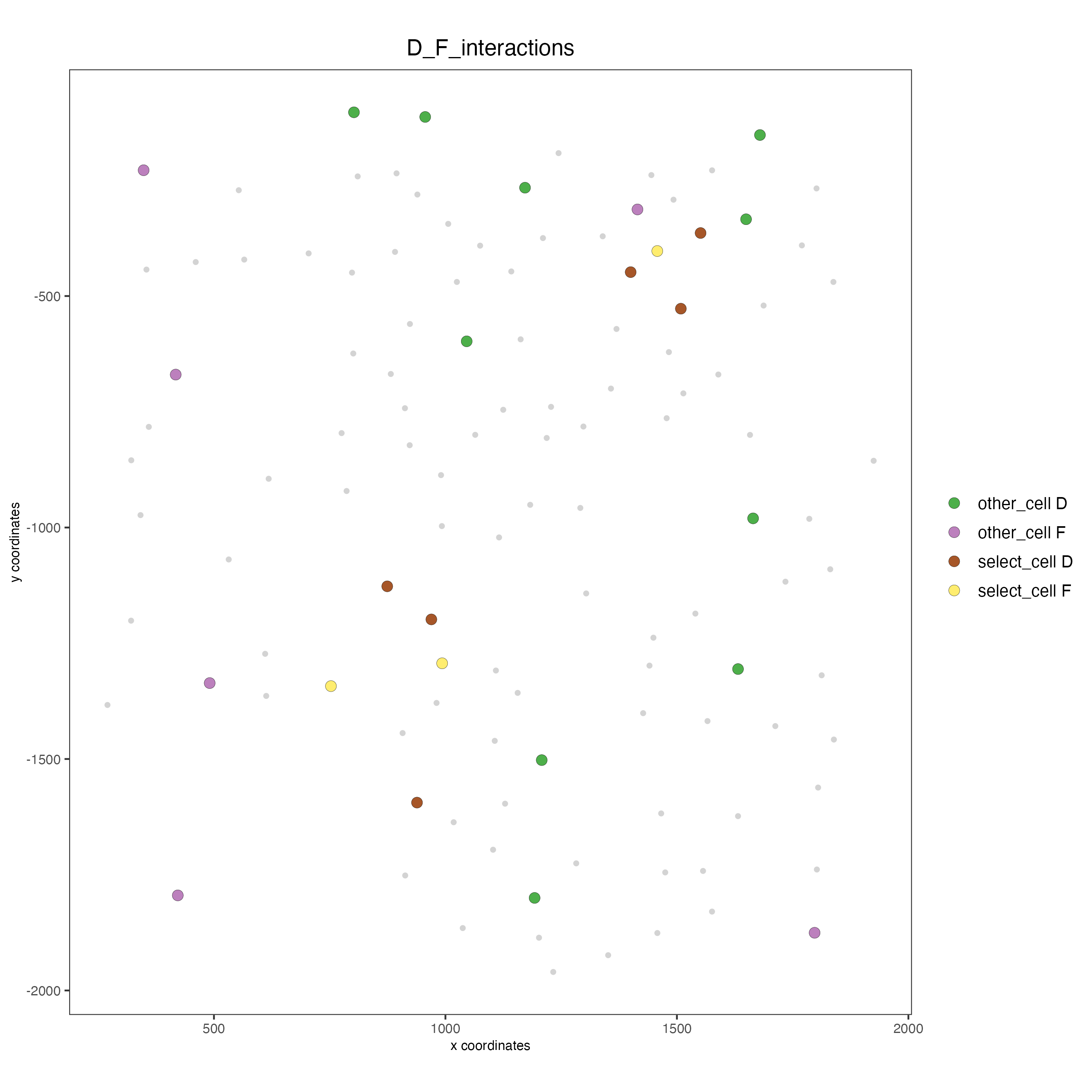
14 Cell neighborhood: Interaction Changed Features
## select top 25 highest expressing genes
gene_metadata <- fDataDT(seqfish_mini)
plot(gene_metadata$nr_cells, gene_metadata$mean_expr)
plot(gene_metadata$nr_cells, gene_metadata$mean_expr_det)
quantile(gene_metadata$mean_expr_det)
high_expressed_genes <- gene_metadata[mean_expr_det > 4]$feat_ID
## identify features (genes) that are associated with proximity to other cell types
ICFscoresHighGenes <- findInteractionChangedFeats(gobject = seqfish_mini,
selected_feats = high_expressed_genes,
spatial_network_name = "Delaunay_network",
cluster_column = "cell_types",
diff_test = "permutation",
adjust_method = "fdr",
nr_permutations = 500,
do_parallel = TRUE)
## visualize all genes
plotCellProximityFeats(seqfish_mini,
icfObject = ICFscoresHighGenes,
method = "dotplot")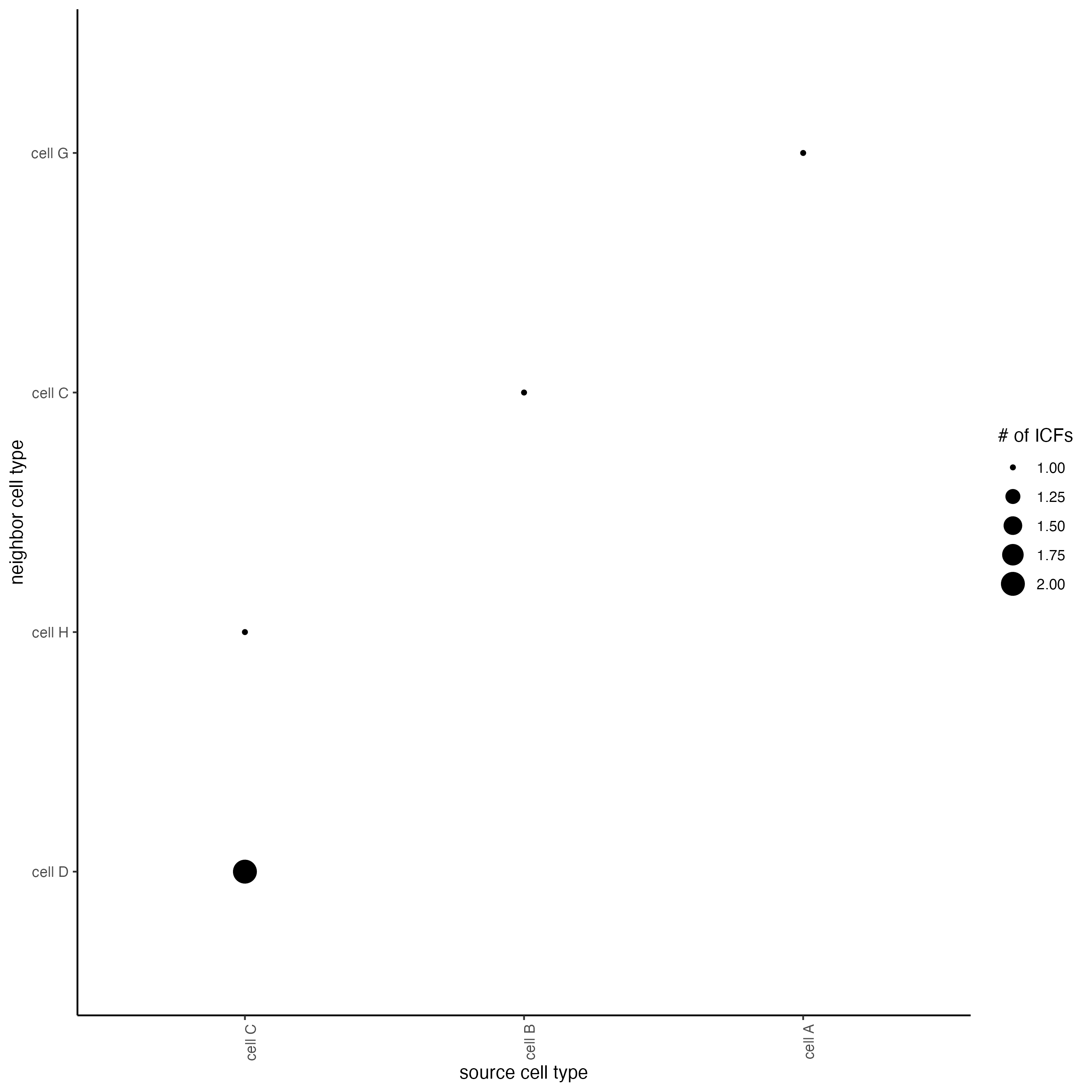
## filter genes
ICFscoresFilt <- filterICF(ICFscoresHighGenes,
min_cells = 2,
min_int_cells = 2,
min_fdr = 0.1,
min_spat_diff = 0.1,
min_log2_fc = 0.1,
min_zscore = 1)
## visualize subset of interaction changed genes (ICGs)
ICF_genes <- c("Cpne2", "Scg3", "Cmtm3", "Cplx1", "Lingo1")
ICF_genes_types <- c("cell E", "cell D", "cell D", "cell G", "cell E")
names(ICF_genes) <- ICF_genes_types
plotICF(gobject = seqfish_mini,
icfObject = ICFscoresHighGenes,
source_type = "cell A",
source_markers = c("Csf1r", "Laptm5"),
ICF_feats = ICF_genes)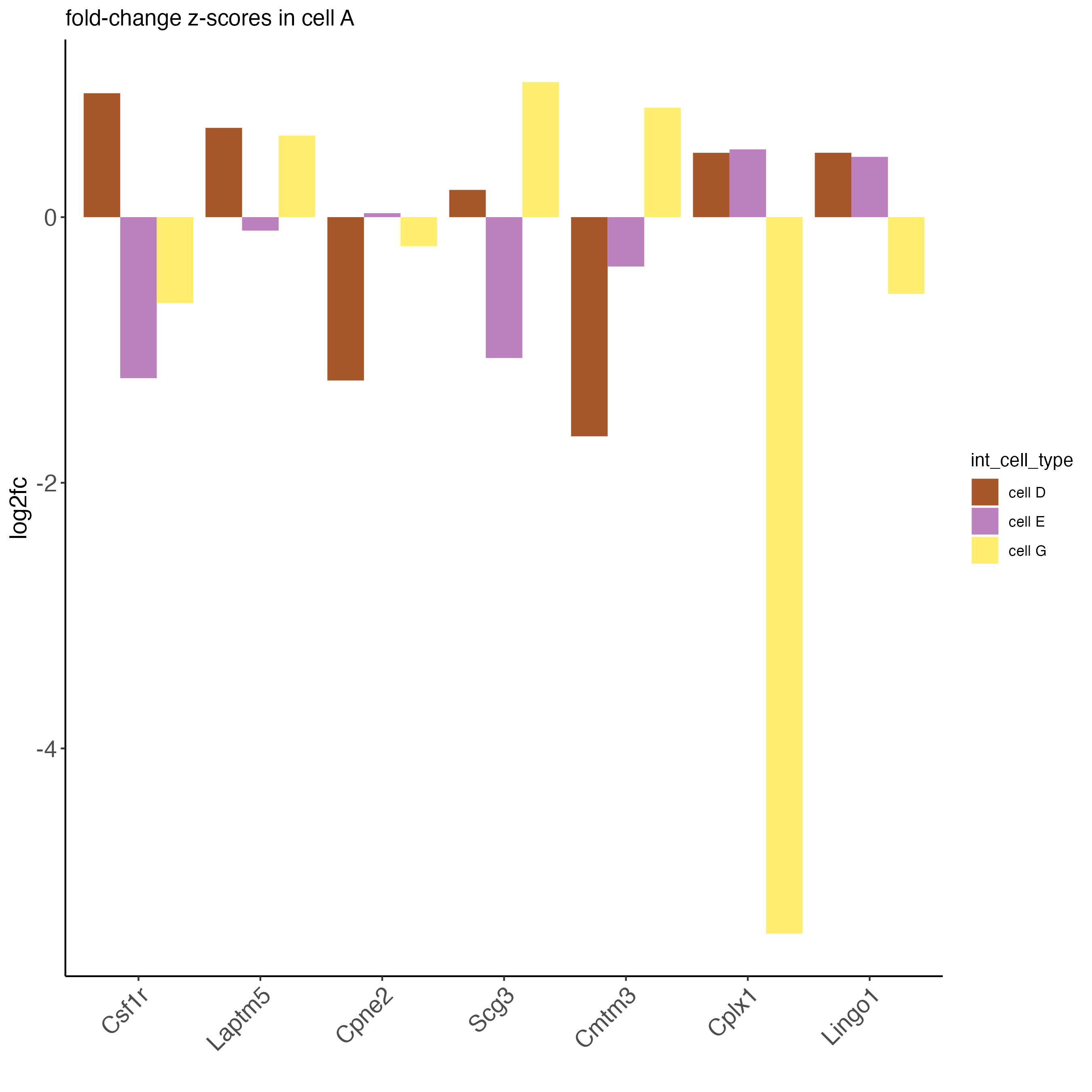
15 Cell Neighborhood: Ligand-Receptor Cell-Cell Communication
LR_data <- data.table::fread(system.file("Mini_datasets/seqfish/Raw/mouse_ligand_receptors.txt",
package = "GiottoData"))
LR_data[, ligand_det := ifelse(mouseLigand %in% seqfish_mini@feat_ID[["rna"]], TRUE, FALSE)]
LR_data[, receptor_det := ifelse(mouseReceptor %in% seqfish_mini@feat_ID[["rna"]], TRUE, FALSE)]
LR_data_det <- LR_data[ligand_det == TRUE & receptor_det == TRUE]
select_ligands <- LR_data_det$mouseLigand
select_receptors <- LR_data_det$mouseReceptor
## get statistical significance of gene pair expression changes based on expression ##
expr_only_scores <- exprCellCellcom(gobject = seqfish_mini,
cluster_column = "cell_types",
random_iter = 50,
feat_set_1 = select_ligands,
feat_set_2 = select_receptors)
## get statistical significance of gene pair expression changes upon cell-cell interaction
spatial_all_scores <- spatCellCellcom(seqfish_mini,
spat_unit = "cell",
feat_type = "rna",
spatial_network_name = "Delaunay_network",
cluster_column = "cell_types",
random_iter = 50,
feat_set_1 = select_ligands,
feat_set_2 = select_receptors,
adjust_method = "fdr",
do_parallel = TRUE,
cores = 4,
verbose = "none")
## * plot communication scores ####
## select top LR ##
selected_spat <- spatial_all_scores[p.adj <= 0.5 & abs(log2fc) > 0.1 & lig_nr >= 2 & rec_nr >= 2]
data.table::setorder(selected_spat, -PI)
top_LR_ints <- unique(selected_spat[order(-abs(PI))]$LR_comb)[1:33]
top_LR_cell_ints <- unique(selected_spat[order(-abs(PI))]$LR_cell_comb)[1:33]
plotCCcomHeatmap(gobject = seqfish_mini,
comScores = spatial_all_scores,
selected_LR = top_LR_ints,
selected_cell_LR = top_LR_cell_ints,
show = "LR_expr")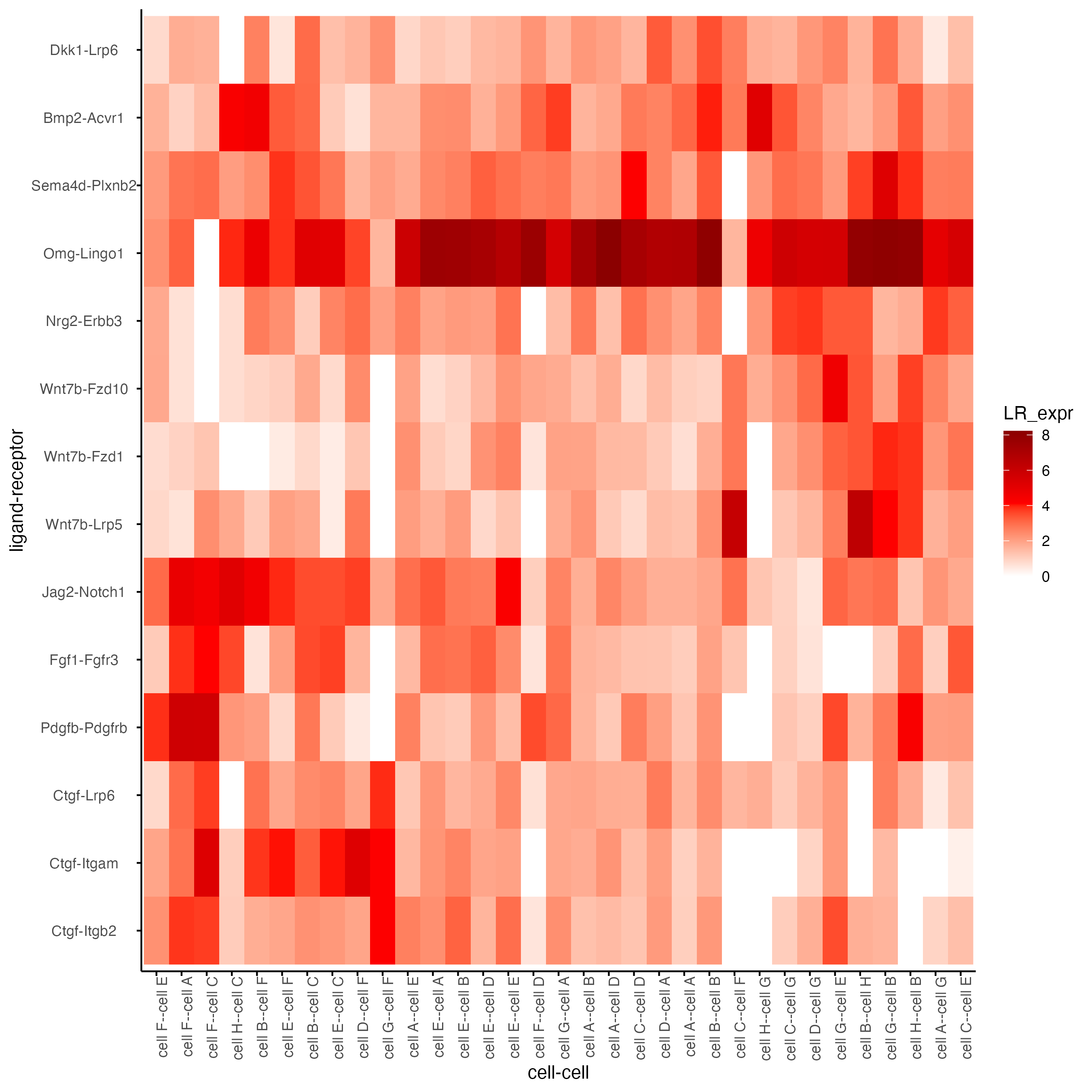
plotCCcomDotplot(gobject = seqfish_mini,
comScores = spatial_all_scores,
selected_LR = top_LR_ints,
selected_cell_LR = top_LR_cell_ints,
cluster_on = "PI")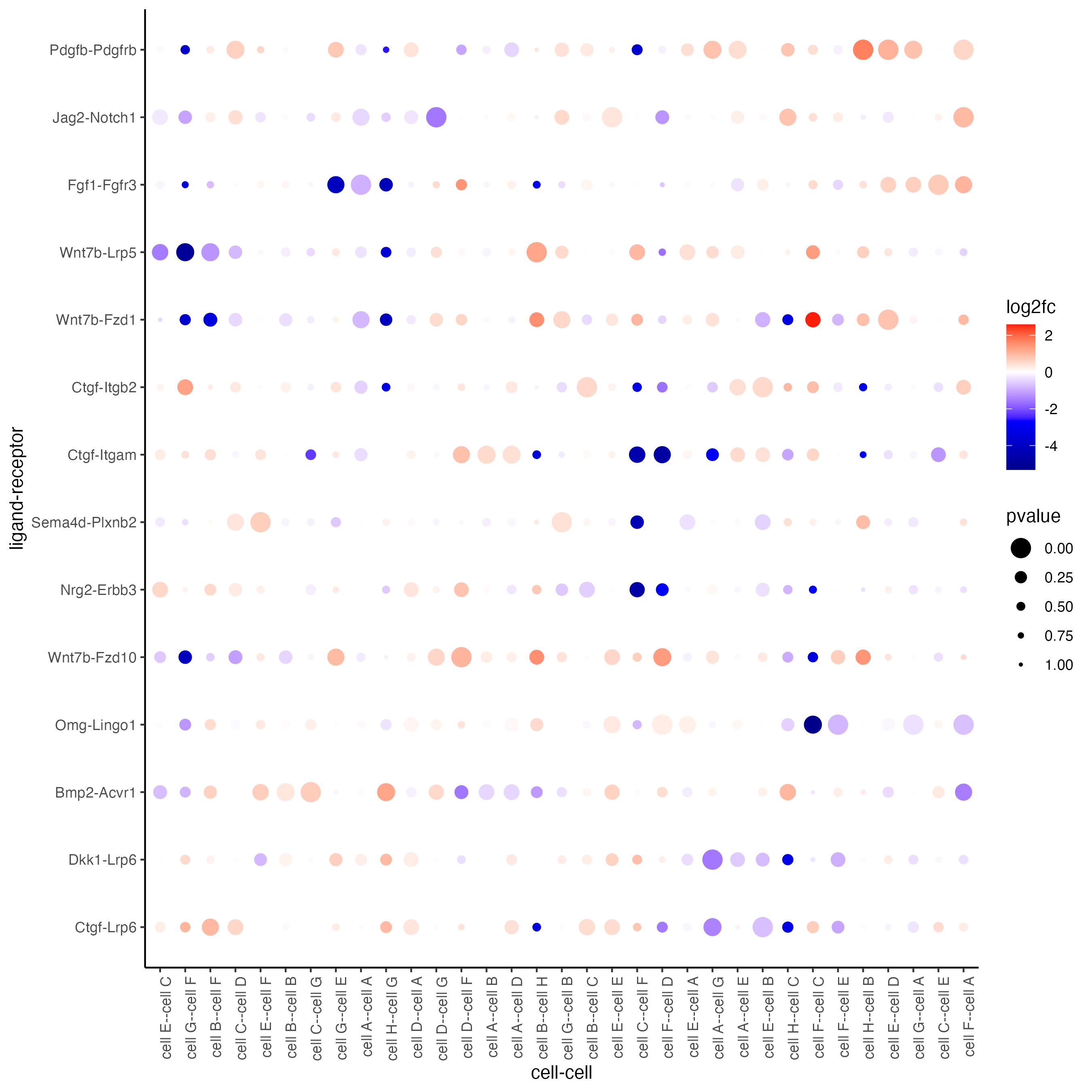
## * spatial vs rank ####
comb_comm <- combCCcom(spatialCC = spatial_all_scores,
exprCC = expr_only_scores)
# top differential activity levels for ligand receptor pairs
plotRankSpatvsExpr(gobject = seqfish_mini,
comb_comm,
expr_rnk_column = "exprPI_rnk",
spat_rnk_column = "spatPI_rnk",
gradient_midpoint = 10)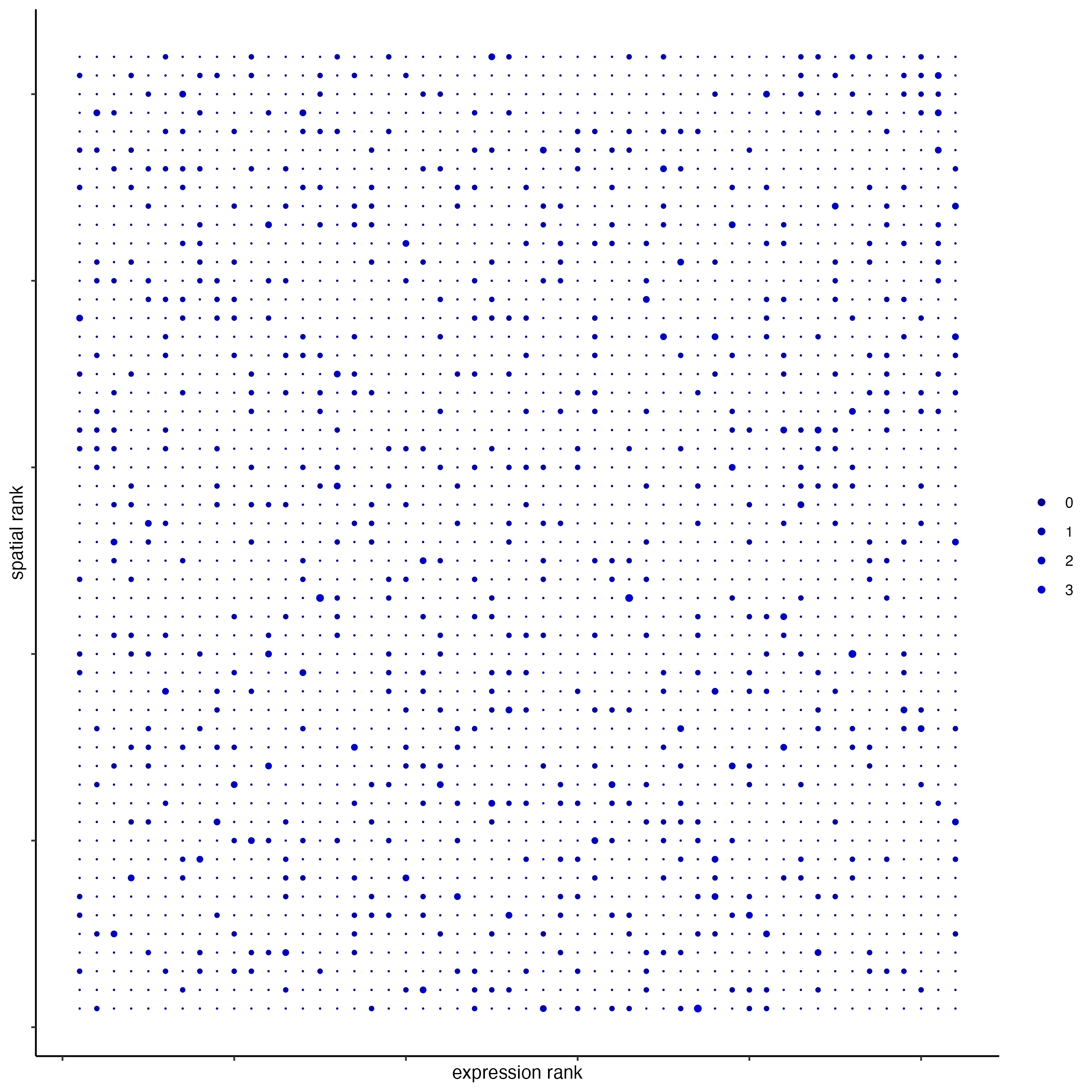
## * recovery ####
## predict maximum differential activity
plotRecovery(gobject = seqfish_mini,
comb_comm,
expr_rnk_column = "exprPI_rnk",
spat_rnk_column = "spatPI_rnk",
ground_truth = "spatial")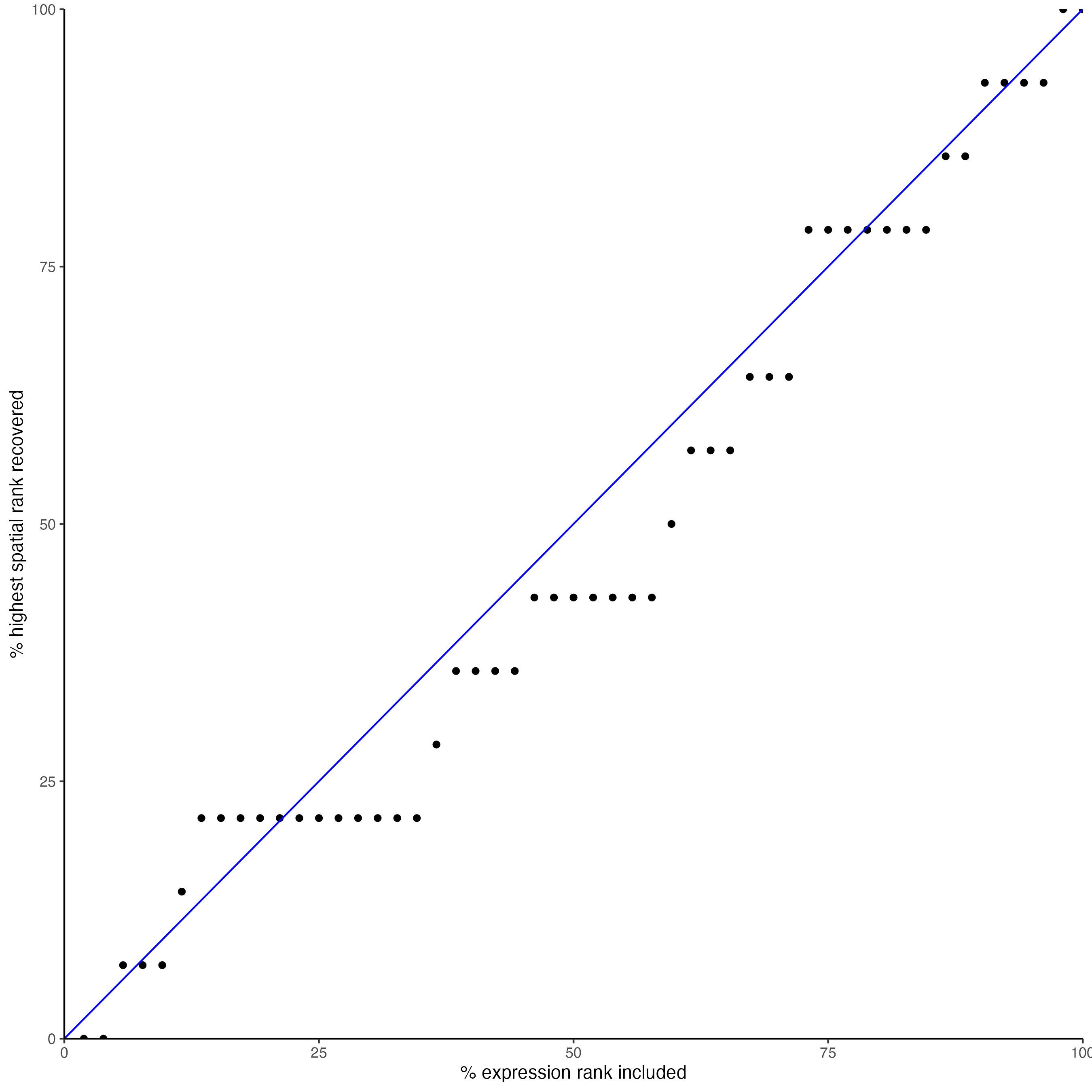
16 Session info
R version 4.3.2 (2023-10-31)
Platform: x86_64-apple-darwin20 (64-bit)
Running under: macOS Sonoma 14.3.1
Matrix products: default
BLAS: /System/Library/Frameworks/Accelerate.framework/Versions/A/Frameworks/vecLib.framework/Versions/A/libBLAS.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.3-x86_64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: America/New_York
tzcode source: internal
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] smfishHmrf_0.1 fs_1.6.3 pracma_2.4.4 ggdendro_0.1.23 GiottoData_0.2.7.0
[6] GiottoUtils_0.1.5 Giotto_4.0.2 GiottoClass_0.1.3
loaded via a namespace (and not attached):
[1] RColorBrewer_1.1-3 rstudioapi_0.15.0 jsonlite_1.8.8
[4] shape_1.4.6 magrittr_2.0.3 magick_2.8.2
[7] farver_2.1.1 rmarkdown_2.25 GlobalOptions_0.1.2
[10] zlibbioc_1.48.0 ragg_1.2.7 vctrs_0.6.5
[13] Cairo_1.6-2 RCurl_1.98-1.14 terra_1.7-71
[16] htmltools_0.5.7 S4Arrays_1.2.0 SparseArray_1.2.4
[19] parallelly_1.36.0 plyr_1.8.9 igraph_2.0.1.1
[22] lifecycle_1.0.4 iterators_1.0.14 pkgconfig_2.0.3
[25] rsvd_1.0.5 Matrix_1.6-5 R6_2.5.1
[28] fastmap_1.1.1 GenomeInfoDbData_1.2.11 rbibutils_2.2.16
[31] MatrixGenerics_1.14.0 future_1.33.1 clue_0.3-65
[34] digest_0.6.34 colorspace_2.1-0 S4Vectors_0.40.2
[37] irlba_2.3.5.1 textshaping_0.3.7 GenomicRanges_1.54.1
[40] beachmat_2.18.0 labeling_0.4.3 progressr_0.14.0
[43] fansi_1.0.6 polyclip_1.10-6 abind_1.4-5
[46] compiler_4.3.2 withr_3.0.0 doParallel_1.0.17
[49] backports_1.4.1 BiocParallel_1.36.0 viridis_0.6.5
[52] ggforce_0.4.1 MASS_7.3-60.0.1 DelayedArray_0.28.0
[55] rjson_0.2.21 gtools_3.9.5 GiottoVisuals_0.1.4
[58] tools_4.3.2 future.apply_1.11.1 glue_1.7.0
[61] dbscan_1.1-12 grid_4.3.2 checkmate_2.3.1
[64] Rtsne_0.17 cluster_2.1.6 reshape2_1.4.4
[67] generics_0.1.3 gtable_0.3.4 tidyr_1.3.1
[70] data.table_1.15.0 tidygraph_1.3.1 BiocSingular_1.18.0
[73] ScaledMatrix_1.10.0 utf8_1.2.4 XVector_0.42.0
[76] BiocGenerics_0.48.1 ggrepel_0.9.5 foreach_1.5.2
[79] pillar_1.9.0 stringr_1.5.1 limma_3.58.1
[82] circlize_0.4.15 dplyr_1.1.4 tweenr_2.0.2
[85] lattice_0.22-5 FNN_1.1.4 deldir_2.0-2
[88] tidyselect_1.2.0 ComplexHeatmap_2.18.0 SingleCellExperiment_1.24.0
[91] knitr_1.45 gridExtra_2.3 IRanges_2.36.0
[94] SummarizedExperiment_1.32.0 stats4_4.3.2 xfun_0.42
[97] graphlayouts_1.1.0 Biobase_2.62.0 statmod_1.5.0
[100] matrixStats_1.2.0 stringi_1.8.3 yaml_2.3.8
[103] evaluate_0.23 codetools_0.2-19 ggraph_2.1.0
[106] tibble_3.2.1 colorRamp2_0.1.0 cli_3.6.2
[109] uwot_0.1.16 reticulate_1.35.0 systemfonts_1.0.5
[112] Rdpack_2.6 munsell_0.5.0 Rcpp_1.0.12
[115] GenomeInfoDb_1.38.6 globals_0.16.2 png_0.1-8
[118] parallel_4.3.2 ggplot2_3.4.4 bitops_1.0-7
[121] listenv_0.9.1 SpatialExperiment_1.12.0 viridisLite_0.4.2
[124] scales_1.3.0 purrr_1.0.2 crayon_1.5.2
[127] GetoptLong_1.0.5 rlang_1.1.3 cowplot_1.1.3