1 Setup and load example dataset
# Ensure Giotto Suite is installed
if(!"Giotto" %in% installed.packages()) {
pak::pkg_install("drieslab/Giotto")
}
# Ensure Giotto Data is installed
if(!"GiottoData" %in% installed.packages()) {
pak::pkg_install("drieslab/GiottoData")
}
library(Giotto)
# Ensure the Python environment for Giotto has been installed
genv_exists <- checkGiottoEnvironment()
if(!genv_exists){
# The following command need only be run once to install the Giotto environment
installGiottoEnvironment()
}
# load the object
g <- GiottoData::loadGiottoMini("visium")2 Gini markers
The Gini method identifies genes that are very selectively expressed in a specific cluster, however not always expressed in all cells of that cluster. In other words, highly specific but not necessarily sensitive at the single-cell level.
- Calculate the top marker genes per cluster using the gini method.
gini_markers <- findMarkers_one_vs_all(gobject = g,
method = "gini",
expression_values = "normalized",
cluster_column = "leiden_clus",
min_feats = 5)
topgenes_gini <- gini_markers[, head(.SD, 2), by = "cluster"]$feats- Visualize
Plot the normalized expression distribution of the top expressed genes.
violinPlot(g,
feats = unique(topgenes_gini),
cluster_column = "leiden_clus",
strip_text = 6,
strip_position = "right")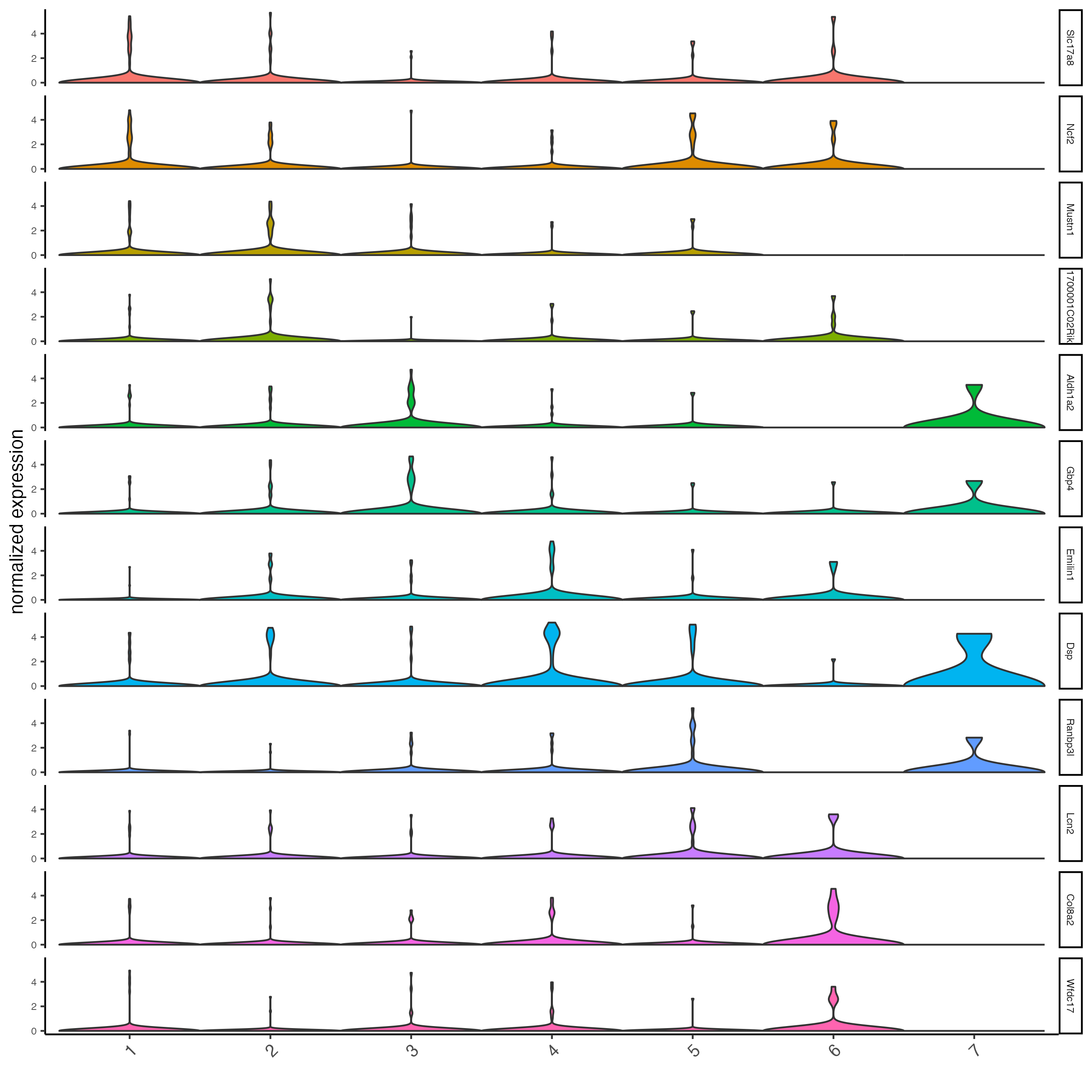
Use the cluster IDs to create a heatmap with the normalized expression of the top expressed genes per cluster.
plotMetaDataHeatmap(g,
selected_feats = unique(topgenes_gini),
metadata_cols = "leiden_clus",
x_text_size = 10, y_text_size = 10)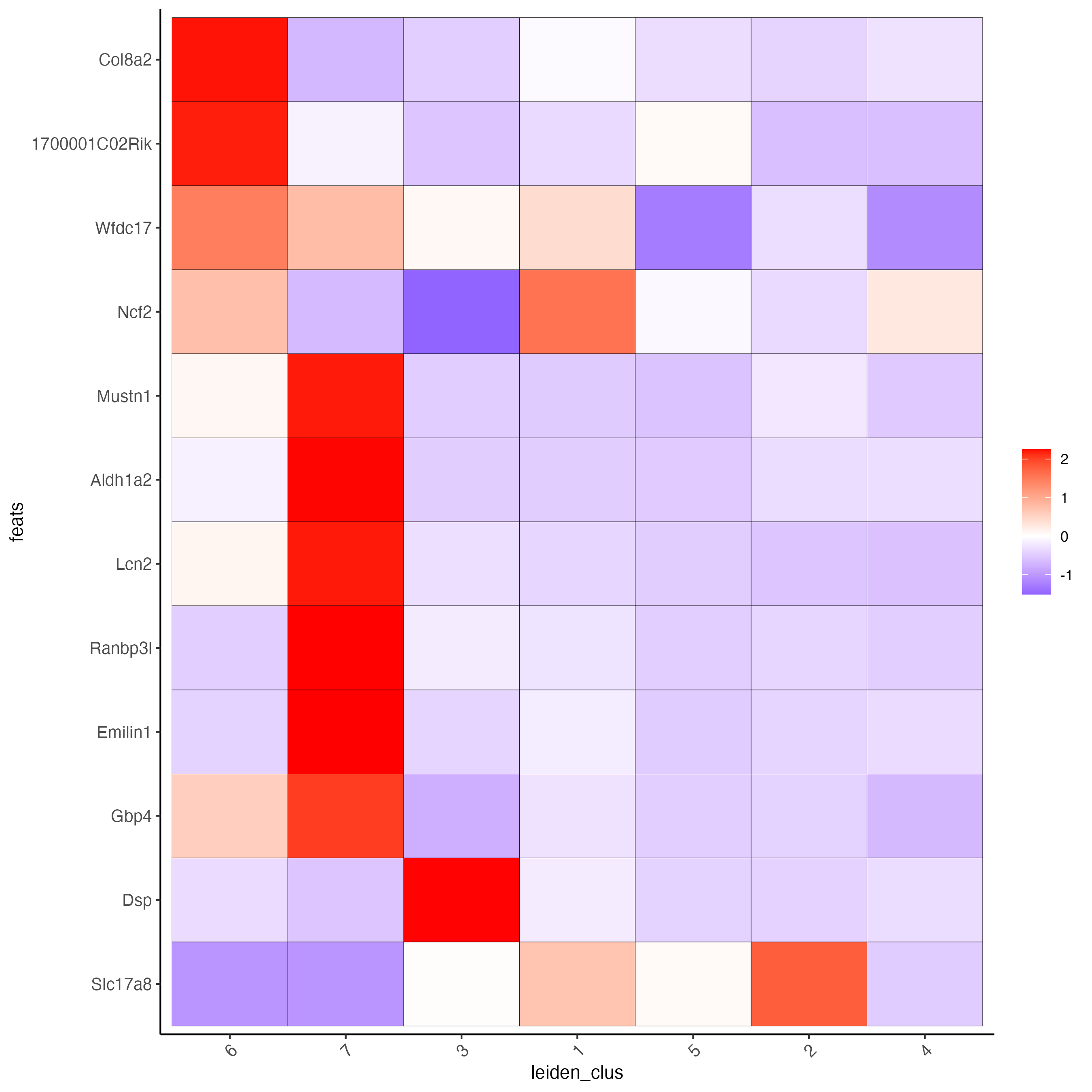
Visualize the scaled expression spatial distribution of the top expressed genes across the sample.
dimFeatPlot2D(g,
expression_values = "scaled",
feats = sort(unique(topgenes_gini)),
cow_n_col = 4,
point_size = 2,
save_param = list(base_width = 20, base_height = 20))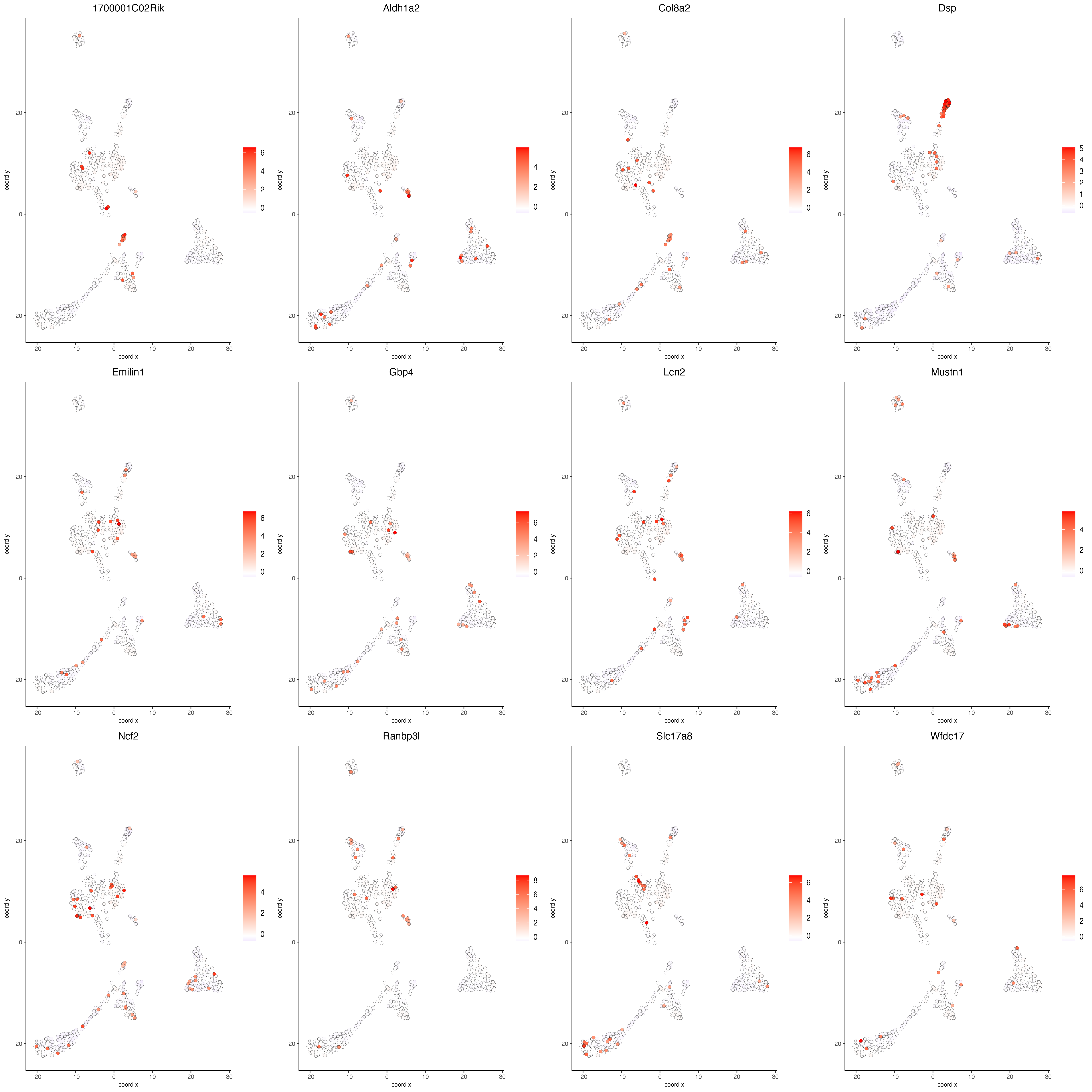
3 Scran markers
The Scran method is preferred for robust differential expression analysis, especially when addressing technical variability or differences in sequencing depth across the sample.
- Calculate the top marker genes per cluster using the scran method
scran_markers <- findMarkers_one_vs_all(gobject = g,
method = "scran",
expression_values = "normalized",
cluster_column = "leiden_clus",
min_feats = 5)
topgenes_scran <- scran_markers[, head(.SD, 2), by = "cluster"]$feats- Visualize
Plot the normalized expression distribution of the top expressed genes.
violinPlot(g,
feats = unique(topgenes_scran),
cluster_column = "leiden_clus",
strip_text = 6,
strip_position = "right")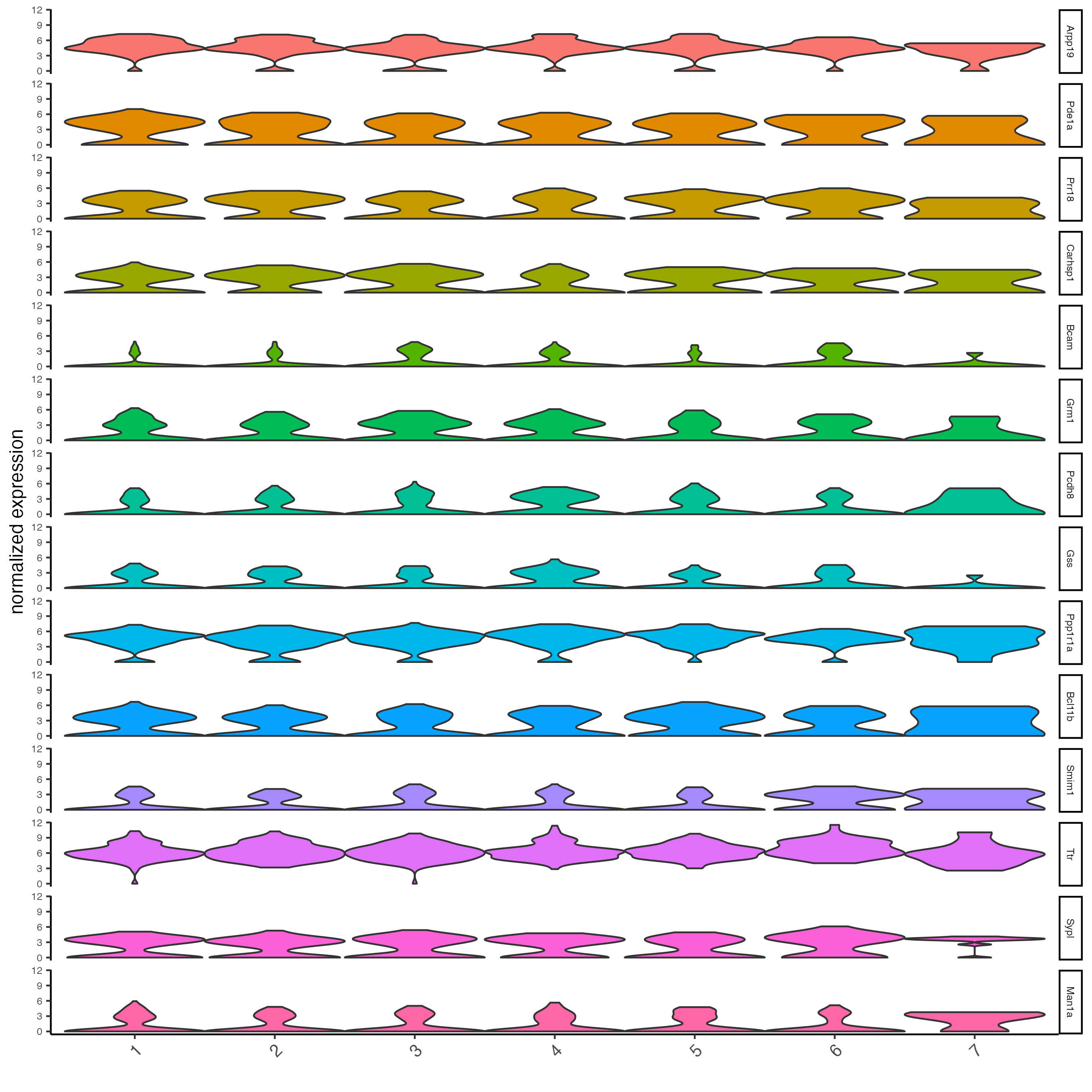
Use the cluster IDs to create a heatmap with the normalized expression of the top expressed genes per cluster.
plotMetaDataHeatmap(g,
selected_feats = unique(topgenes_scran),
metadata_cols = "leiden_clus",
x_text_size = 10, y_text_size = 10)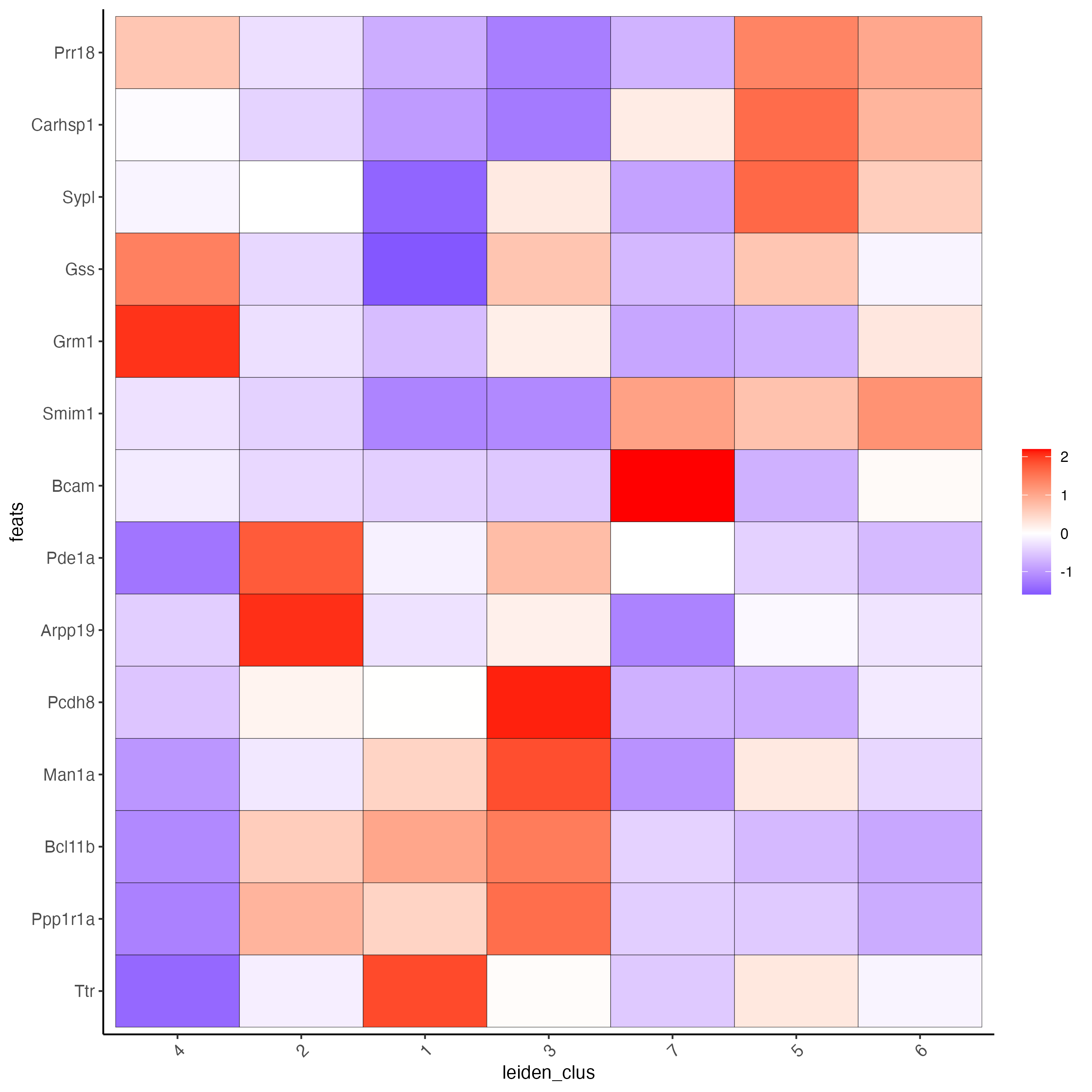
Visualize the scaled expression spatial distribution of the top expressed genes across the sample.
dimFeatPlot2D(g,
expression_values = "scaled",
feats = sort(unique(topgenes_scran)),
cow_n_col = 4,
point_size = 1,
save_param = list(base_width = 20, base_height = 20))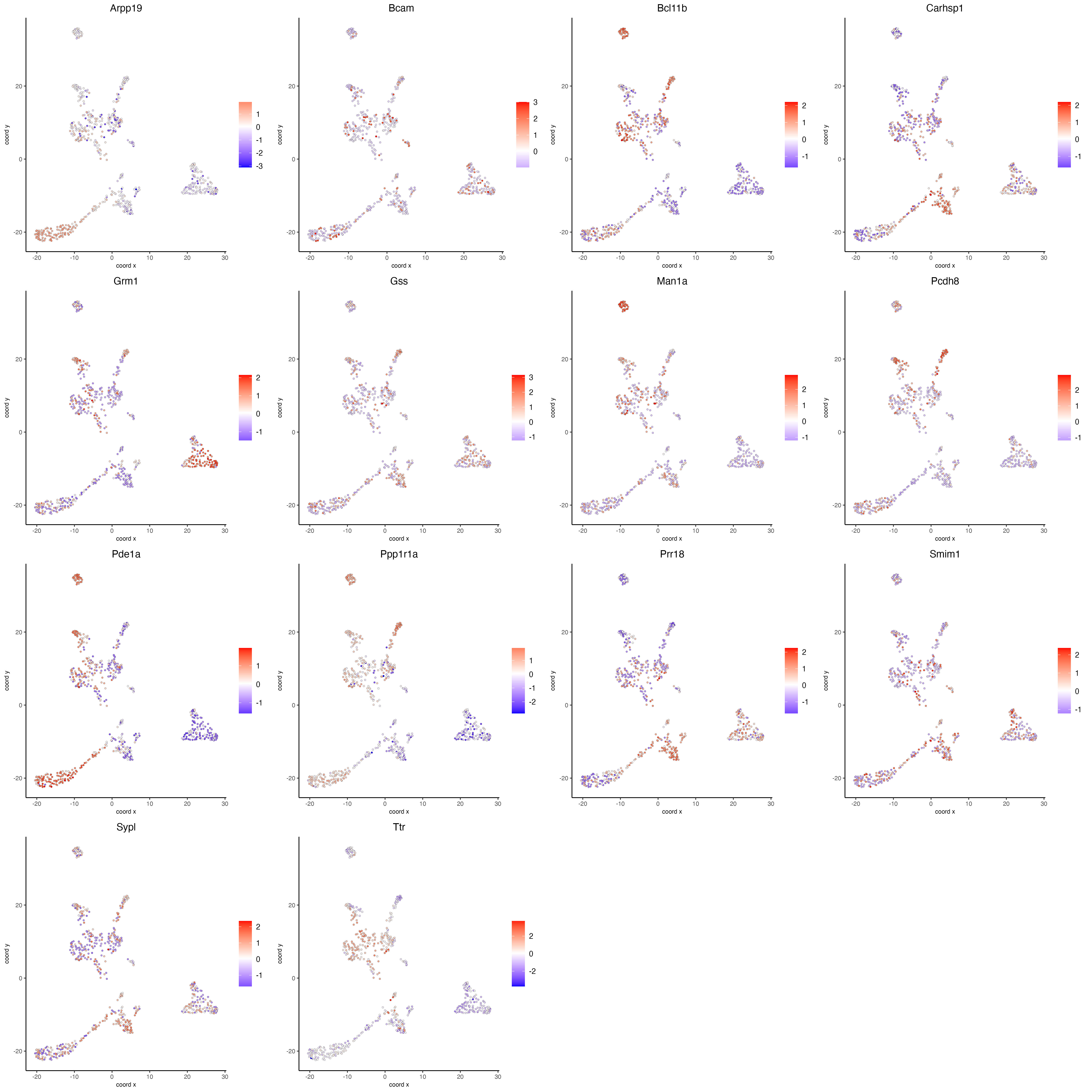
In practice, it is often beneficial to apply both Gini and Scran methods and compare results for a more complete understanding of differential gene expression across clusters.
4 Session info
R version 4.4.2 (2024-10-31)
Platform: x86_64-apple-darwin20
Running under: macOS Sequoia 15.0.1
Matrix products: default
BLAS: /System/Library/Frameworks/Accelerate.framework/Versions/A/Frameworks/vecLib.framework/Versions/A/libBLAS.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.4-x86_64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: America/New_York
tzcode source: internal
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] Giotto_4.1.4 GiottoClass_0.4.3
loaded via a namespace (and not attached):
[1] colorRamp2_0.1.0 rlang_1.1.4
[3] magrittr_2.0.3 GiottoUtils_0.2.0
[5] matrixStats_1.4.1 compiler_4.4.2
[7] DelayedMatrixStats_1.26.0 png_0.1-8
[9] systemfonts_1.1.0 vctrs_0.6.5
[11] pkgconfig_2.0.3 SpatialExperiment_1.14.0
[13] crayon_1.5.3 fastmap_1.2.0
[15] backports_1.5.0 magick_2.8.5
[17] XVector_0.44.0 scuttle_1.14.0
[19] labeling_0.4.3 utf8_1.2.4
[21] rmarkdown_2.28 UCSC.utils_1.0.0
[23] ragg_1.3.3 purrr_1.0.2
[25] xfun_0.47 bluster_1.14.0
[27] zlibbioc_1.50.0 beachmat_2.20.0
[29] GenomeInfoDb_1.40.1 jsonlite_1.8.9
[31] DelayedArray_0.30.1 BiocParallel_1.38.0
[33] terra_1.7-78 cluster_2.1.6
[35] irlba_2.3.5.1 parallel_4.4.2
[37] R6_2.5.1 limma_3.60.4
[39] reticulate_1.39.0 GenomicRanges_1.56.1
[41] scattermore_1.2 Rcpp_1.0.13
[43] SummarizedExperiment_1.34.0 knitr_1.48
[45] IRanges_2.38.1 Matrix_1.7-1
[47] igraph_2.0.3 tidyselect_1.2.1
[49] rstudioapi_0.16.0 abind_1.4-8
[51] yaml_2.3.10 codetools_0.2-20
[53] lattice_0.22-6 tibble_3.2.1
[55] Biobase_2.64.0 withr_3.0.1
[57] evaluate_1.0.0 pillar_1.9.0
[59] MatrixGenerics_1.16.0 checkmate_2.3.2
[61] stats4_4.4.2 plotly_4.10.4
[63] generics_0.1.3 S4Vectors_0.42.1
[65] ggplot2_3.5.1 sparseMatrixStats_1.16.0
[67] munsell_0.5.1 scales_1.3.0
[69] GiottoData_0.2.15 gtools_3.9.5
[71] glue_1.8.0 metapod_1.12.0
[73] lazyeval_0.2.2 tools_4.4.2
[75] GiottoVisuals_0.2.7 BiocNeighbors_1.22.0
[77] data.table_1.16.0 ScaledMatrix_1.12.0
[79] locfit_1.5-9.10 scran_1.32.0
[81] cowplot_1.1.3 grid_4.4.2
[83] tidyr_1.3.1 edgeR_4.2.1
[85] colorspace_2.1-1 SingleCellExperiment_1.26.0
[87] GenomeInfoDbData_1.2.12 BiocSingular_1.20.0
[89] cli_3.6.3 rsvd_1.0.5
[91] textshaping_0.4.0 fansi_1.0.6
[93] S4Arrays_1.4.1 viridisLite_0.4.2
[95] dplyr_1.1.4 gtable_0.3.5
[97] digest_0.6.37 progressr_0.14.0
[99] BiocGenerics_0.50.0 dqrng_0.4.1
[101] SparseArray_1.4.8 ggrepel_0.9.6
[103] rjson_0.2.23 htmlwidgets_1.6.4
[105] farver_2.1.2 htmltools_0.5.8.1
[107] lifecycle_1.0.4 httr_1.4.7
[109] statmod_1.5.0